
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Синдром Лея - клиника, диагностика
Митохондриальные формы атаксий могут развиваться не только при повреждении собственно митохондриальной фракции ДНК, но и в случаях мутаций ядерных генов, отвечающих за синтез разнообразных митохондриальных белков.
Классическим примером ядерно-кодируемой митохондриальной атаксии является атаксия Фридрейха, которая в силу своей распространенности и значимости подробно рассмотрена в самостоятельном разделе монографии. Ниже кратко представлены другие, более редкие формы митохондриальных атаксий с ядерным (менделевским) наследованием.
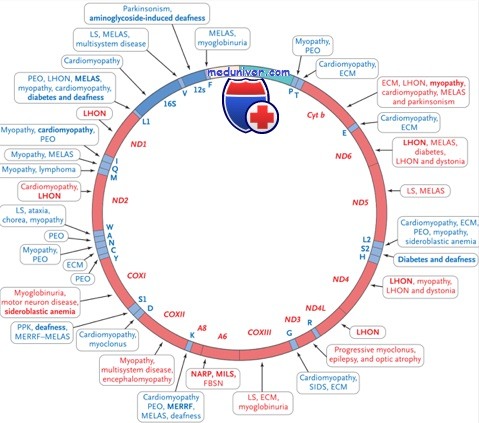
Атаксии, обусловленные дефектами структурных компонентов дыхательной цепи митохондрий. Данная группа заболеваний связана с генетическим повреждением белковых субъединиц отдельных комплексов дыхательной цепи, что сопровождается обычно развитием тяжелого симптомокомплекса — подострой некротизирующей энцефаломиелопатии, или синдрома Лея.
Синдром Лея (MIM 256000) является одной из наиболее частых форм митохондриальных болезней дыхательной цепи у пациентов младенческого или раннего детского возраста. Заболевание развивается на фоне резкой задержки психомоторного развития ребенка и проявляется дыхательными нарушениями, экстрапирамидными симптомами, атаксией, дискоординацией движений глазных яблок, спастичностью, атрофией зрительных нервов, а также подострыми эпизодами тяжелой клинико-биохимической декомпенсации на фоне интеркуррентных инфекций.
Весьма характерен лактат-ацидоз, особенно в период обострения симптоматики. Продолжительность жизни обычно не превышает 5 лет. Типичные изменения в веществе мозга — билатеральные некрозы в области зрительного бугра, базальных ганглиев, ствола мозга, мозжечка (вовлекаются обычно зубчатые ядра и окружающее их белое вещество), а также диффузная демиелинизация и глиоз. Феномен «рваных красных волокон» не характерен.
Синдром Лея — генетически гетерогенное состояние. В ряде случаев он может быть связан с мутациями мтДНК (Chinnery P. et al., тогда как у значительной части больных синдром Лея имеет аутосомно-рецессивное наследование и обусловлен многочисленными мутациями ядерных генов, кодирующих различные белковые субъединицы комплексов I, II и IV дыхательной цепи.
При этом биохимически определяется дефицит соответствующих ферментов митохондрий (Bertini Е. et al.). Среди многообразия молекулярных дефектов следует особо отметить ядерный ген SURF-1, мутированный у абсолютного большинства больных с синдромом Лея и патологией комплекса IV (цитохром-С-оксидаза). Интересно, что белковый продукт данного гена не является собственно субъединицей цитохром-С-оксидазы, а участвует в сборке комплекса IV на внутренней митохондриальной мембране (Zhu Z. et al.). Специфическое лечение не разработано.
- Читать "Атаксии при нарушениях контроля стабильности митохондриальной ДНК"
Оглавление темы "Митохондриальные атаксии":- Нейросенсорная тугоухость, атаксия и миоклонус - клиника, диагностика
- Синдром Кернса—Сэйра - клиника, диагностика
- Лечение митохондриальных атаксий
- Синдром Лея - клиника, диагностика
- Атаксии при нарушениях контроля стабильности митохондриальной ДНК
- Атаксии при нарушениях утилизации митохондриальных субстратов
- Церебротендинальный ксантоматоз (ксантоматоз мозга и сухожилий) - клиника, диагностика
- Болезнь Ниманна—Пика - клиника, диагностика
- Абеталипопротеинемия (синдром Бассена—Корнцвейга) - клиника, диагностика
- Атаксия при GM2-ганглиозидозе - клиника, диагностика