
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Несовершенное костеобразование (НК) - фенотип, диагностика
Несовершенное костеобразование при первичном осмотре больного ребенка и выслушивании жалоб родителей, как нам кажется, не представляет особых трудностей для диагностики. Когда врач осматривает ребенка с грубыми скелетными деформациями, наступившими в результате спонтанных переломов длинныхтрубчатых костей, он понимает, что речь идет о наследственном заболевании скелета - несовершенном костеобразовании или osteogenesis imperfecta.
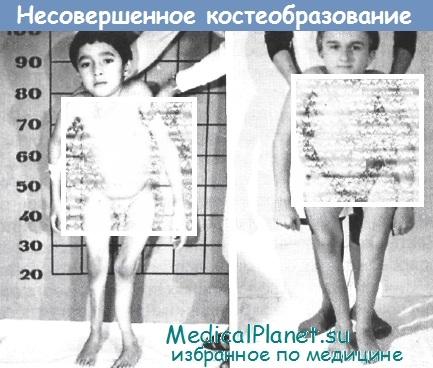
На фотографиях видна вся гамма разнообразных скелетных аномалий у детей разного возраста. Они свидетельствуют о тяжелой инвалидности. Причем у части из них переломы наступали внутриутробно или вскоре после рождения, а их количество могло достигать 15-55. В результате часто повторявшихся переломов у детей возникли саблевидные деформации голеней, варусная деформация бедренных костей, искривление костей верхних конечностей, килевидная деформация грудной клетки, сколиоз или кифосколиоз.
При этом интеллектуальное развитие детей соответствовало возрасту (IQ - 85-90 ед.). И это особенно драматизирует ситуацию. Маленькие дети имеют более зрелый интеллект, они понимают безнадежность своего состояния, их очевидная инвалидность (на фоне других детей) обостряет восприятие окружающего мира и своего места в нем. Они не могут самостоятельно передвигаться, каждое движение нередко сопровождается болями, неловкие движения грозят очередными переломами.
При анализе родословных этих детей часто встречаются спорадические случаи возникновения заболевания. В иных родословных аналогичные заболевания или отдельные симптомы несовершенного остеогенеза (тугоухость, голубые склеры и др.) часто обнаруживаются у ближайших и отдаленных родственников пробанда. Так, например, несовершенное костеобразование (НК) нередко прослеживается в трех поколениях одной семьи и свидетельствует об аутосомно-доминантном типе наследования.
Клиническая картина синдрома определяется многочисленными спонтанно возникающими переломами и последующими деформациями скелета: искривление длинных трубчатых костей, сколиоз, бочкообразная или килевидная грудная клетка; нарушения дентинообразования.
Перечень возможных скелетных аномалий при несовершенном костеобразовании
1. Переломы длинных трубчатых костей:
- Внутриутробные,
- Сразу после рождения,
- Впервые 6 мес,
- После 1 года,
- После 3 лет.
2. Общее количество переломов:
- Более 30,
- 15-25, -до 5.
3. Изменение костей черепа:
- Уплощение в сагиттальном размере,
- Увеличение лобных и теменных бугров,
- Позднее закрытие большого родничка.
4. Вывих плечевого сустава.
5. Последствия костных переломов:
- Деформация костей голени, бедер,
- Деформация костей верхних конечностей,
- Деформация позвоночного столба (кифосколиоз, кифоз),
- Деформация грудной клетки,
- Укорочение нижних конечностей,
- Посттравматическая контрактура.
6. Способность к передвижению:
- Полностью отсутствует,
- С помощью костылей,
- Сохранена.
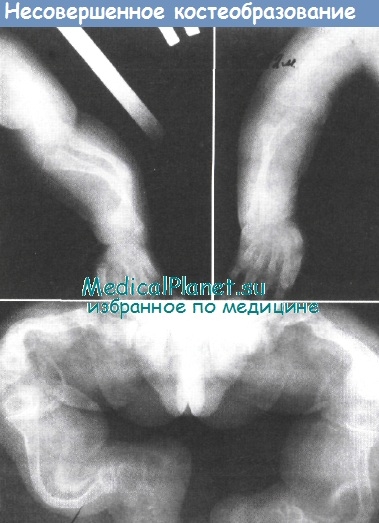
Рентгенологически, как правило, обнаруживают грубые нарушения костных структур:
• остеопороз, в том числе системный и локальный.
• зоны перестройки костной ткани,
• наличие свежих патологических переломов,
• изменение структуры губчатого вещества,
• осевые деформации длинных трубчатых костей,
• эксцентрическая атрофия длинных трубчатых костей,
• расширение метаэпифизарных отделов трубчатых костей,
• замедление роста костей в длину,
• уплощение тела позвонков,
• деформация таза по типу «карточного сердца»,
• истончение костей свода черепа,
• наличие аномалий развития скелета.
При морфологическом изучении биоптатов костной ткани подвздошной кости становится очевидным, насколько грубыми оказываются изменения костной структуры. На первый план выступают явления костной дисплазии в виде сужения или отсутствия зон энхондрального окостенения, очагов пазушного рассасывания и резорбции, повышенного количества остеоцитов.
Исследователи единодушны в том, что несовершенное костеобразование (НК) является гетерогенным заболеванием и работы последних лет убеждают в этом. Однако до сих пор в практических целях принята условная классификация, согласно которой выделяют два главных типа: поздний и врожденный.
Условная классификация несовершенного остеогенеза
I. Поздний.
Аутосомно-доминантное наследование в большинстве случаев; тяжесть течения варьирует; почти несомненна генетическая гетерогенность.
Аутосомно-рецессивный тип, встречается крайне редко; течение тяжелое с прогрессирующей деформацией.
II. Врожденный.
Спорадическая форма, по-видимому, обусловлена новыми доминантными мутациями; подавляющее большинство случаев.
Аутосомно-рецессивный тип; встречается редко; на рентгенограмме кости обычно утолщены, встречается симптом реберных четок.
И тем не менее в последние годы это заболевание привлекло к себе пристальное внимание исследователей, которые считают, что несовершенное костеобразование (НК) является заболеванием, входящим в группу костных дисплазий. Оно генетически гетерогенно и в основе лежат изменения в синтезе проколлагена I.
В настоящее время выделено 7 мутантныхтипов: osteogenesis imperfecta I, II, III, IV, V, VI, VII, отличающихся друг от друга локализацией мутантных генов. Так, например, новый VII тип картирован на коротком плече хромосомы 3. При типе III глицин (Gly 511) замещает серотонин (SER) в коллагеновом гене COLIA1 или COLIA2, кодирующих I тип проколлагеновой цепи. Увеличение интенсивности коллагенового метаболизма в III типе объясняют новой мутацией GS 11S.
Анализируя смертность при разных типах несовершенного костеобразования (НК), авторы пришли к выводу, что особенно тяжелым течением отличается тип II (100% смертность). Существовавшее до сих пор мнение, что НК относительно редкое заболевание претерпевает изменения. Так, в Англии и Уэльсе за последние 13 лет выявлено 743 пациента с НК. Причем по возрасту наступления смертельного исхода выделены 3 группы (тип I А, тип III и тип I В, IVA и IV В), при которых смерть наступала в возрасте 25-34-45 лет.
С новых позиций патогенетические механизмы несовершенного костеобразования (НК) становятся еще более неясными и дискутабельными. До недавнего времени ведущую роль в генезе НК придавали нарушениям соединительной ткани: межклеточному веществу, волокнистым структурам (коллагеновые, ретикулиновые и эластичные волокна, соединительнотканные клетки — фибробласты и тучные клетки).
Естественно, при этом диагностическое значение придавалось определению кислых гликозаминогликанов (ГАГ). Определение фракционного состава ГАГ мочи выявляет преимущественную почечную экскрециюхондроитин-4,6-сульфатов и гепарансульфата, повышение карбазолорцинолового коэффициента. Причем снижение фракций дерматансульфата характерно для тяжело протекающего НК и прогностически расценивается как неблагоприятный признак.
Другими важными компонентами структуры и метаболизма соединительной ткани являются оксипролин и оксилизингликозиды, входящие в состав фибриллярных белков и отражающие интенсивность катаболизма коллагена.
Преобладанием оксилизина пытаются объяснить нарушения процессов минерализации, остеолиза, метаболизма коллагена, врожденную недостаточность остеобластов и др. Детально обсуждается роль паратгормона, кальцитонина, витаминай, гормонов надпочечников. Действительно, удетей с НК не только изменяются основные показатели обмена соединительной ткани, когда почечная экскреция ГАГ и оксипролина превышает возрастную норму в 1,5-4 раза, но и наступает компенсаторное увеличение факторов, регулирующих фосфорно-кальциевый обмен: паратгормона, кальцитонина, кальцидиола и глюкопротеидов.
Таким образом, в генезе несовершенного костеобразования (НК) большая роль отводится также железам внутренней секреции (паращитовидные, щитовидная железа и кора надпочечников), способных оказывать влияние на процессы минерализации кости и минеральный обмен. Наряду с этим существует гипотеза о нарушениях процессов минерализации за счет расстройств энергетического баланса. Таким образом, предположений о генезе НК существует много, но все они имеют свои недостатки и нередко противоречат друг другу.
Вне зависимости от типа заболевания можно выделить общие для всех типов несовершенного костеобразования (НК) признаки (И.М. Новикова):
• дисплазия костной ткани, определяемая по данным морфологического исследования,
• нормальные показатели кальциевого обмена,
• нормальное кишечное всасывание кальция и фосфора,
• повышение концентрации неорганического фосфора в сыворотке крови,
• сниженная реабсорбция фосфатов в почечных канальцах,
• гиперпаратиреодинемия,
• повышение кальцидола в сыворотке крови,
• повышение уровня оксипролина в плазме.
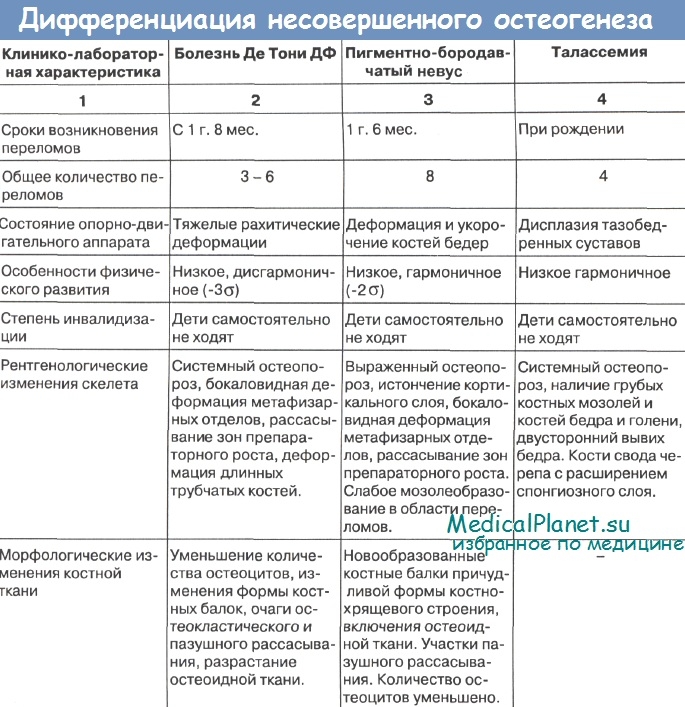
При классической форме несовершенного костеобразования (НК) диагноз не вызывает значительных трудностей. Однако существует целый ряд фенотипически схожих заболеваний, требующих дифференциальной диагностики.
В лечении несовершенного костеобразования (НК) не существует каких-либо методов или лекарств, приводящих к полному выздоровлению. Лечение носит сугубо симптоматический характер: нехирургическое (физиотерапия, климатотерапия и пр), хирургическая коррекция и медикаментозные средства, увеличивающие плотность кости и снижение частоты переломов.
В последние годы в лечении несовершенного костеобразования (НК) многие исследователи пытаются использовать бифосфонат или памидроновую кислоту, применение которой снижает резорбцию костной ткани, оказывает антиостеолитическое и антиостеопорозное действие. В костной ткани эта кислота связывается с кристаллами гидроксиапатита (фосфаты кальция) костной ткани и препятствует их растворению, тормозит пенетрацию в костную ткань предшественников остеобластов и их трансформацию в зрелые формы. Бифосфонаты (обладающие тяжелыми побочными эффектами), тем не менее, все чаще используют при лечении тяжелых форм НК.
Благоприятный эффект от терапии памидронатом на кости и минеральный обмен отмечен при несовершенном костеобразовании (НК) I, III и IV типов у детей в возрасте 8 недель- 18 лет. Утверждают также, что бифосфонаты способствуют более высокой продукции коллагена и уменьшению числа переломов. В процессе лечения памидронатом обязательно осуществляется контроль за метаболизмом: определяется в плазме 1,25-дигидроксихолекальциферол и 25-гидроксихолекальциферол, инсулин-подобный фактор роста-1 (IGF-1), транспортный белок (IGFBP3), остеокальцин, общая щелочная фосфотаза и паратгормон; денситометрия и рентген.
Несмотря на отмечаемые положительные лечебные эффекты памидроната, большинство исследователей признают, что будущее в лечении osteogenesis imperfecta принадлежит генной инженерии.
Для акушеров может представлять интерес, что беременность при НК встречается с частотой 1 : 25 000-1 : 30 000. Родоразрешение преимущественно осуществляется путем кесарева сечения. Послеродовые осложнения сводятся к маточным кровотечениям за счет атонии матки. Сообщается также об успешной пренатальной диагностике НК I типа, при этом была обнаружена новая мутация COL1А1 гена.
- Рекомендуем далее ознакомиться со статьей "Врожденный множественный артрогрипоз (ВМА) - фенотип, диагностика"
Оглавление темы "Наследственные заболевания у детей":- Синдром Марфана - фенотип, диагностика
- Гомоцистинурия - фенотип, диагностика
- Гиперлипопротеидемия (ГЛП) - фенотип, диагностика
- Синдром Кнаппа-Комровера (наследственная витамин В6-зависимая ксантуренурия) - фенотип, диагностика
- Несовершенное костеобразование (НК) - фенотип, диагностика
- Врожденный множественный артрогрипоз (ВМА) - фенотип, диагностика
- Синдром Дауна - фенотип, диагностика
- Основные причины задержки психомоторного развития детей
- Основные причины микроцефалии
- Основные причины макроцефалии