
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Синдром Марфана - фенотип, диагностика
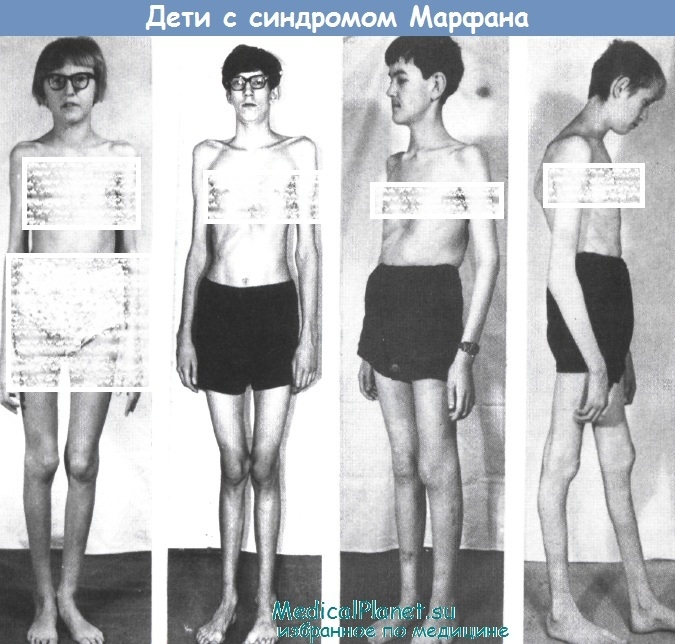
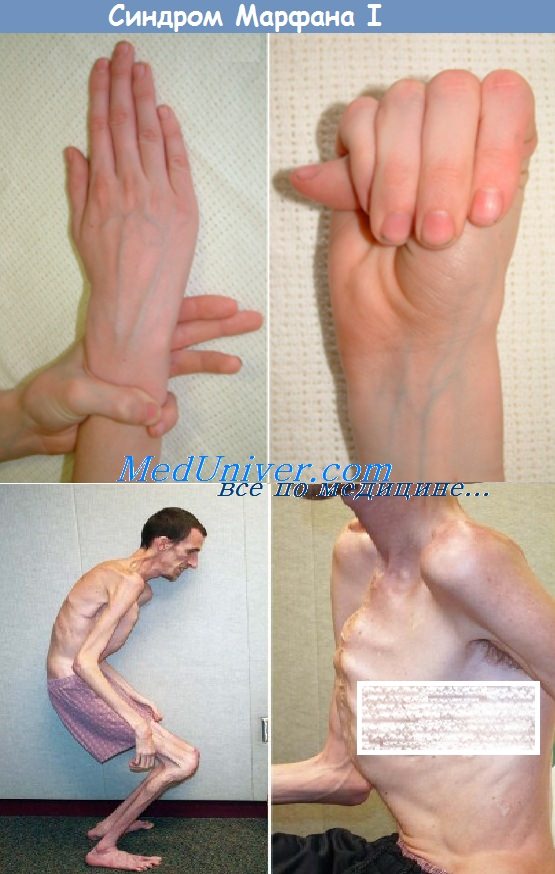
На амбулаторном приеме больной ребенок. При его визуальном осмотре бросается в глаза своеобразие фенотипа: высокий рост, превышающий возрастную норму, астеническое телосложение, скелетные деформации и патология зрения. Среди аномалий скелета: «крыловидные» лопатки, сколиоз или же кифосколиоз, вальгусная деформация нижних конечностей и др. Наряду с этим обнаруживается измененная форма грудной клетки: воронкообразная («грудь сапожника») или килевидная грудь.
Однако одним из главных симптомов является арахнодактилия. Наиболее вероятным окажется, что у ребенка с такими симптомами имеется болезнь Марфана, описанная еще в 1896 г. Арахнодактилия послужила основанием для первичного названия этой болезни.
Помимо перечисленных скелетных аномалий у детей с синдромом Марфана отмечается своеобразное строение лицевого скелета: «птичье» выражение лица, гипертелоризм, антимонголоидный разрез глаз, крупный нос, большие или слишком маленькие уши, низкое их расположение, готическое небо, неправильный прикус и рост зубов.
Синдром Марфана относится к наследственным заболеваниям, связанным с нарушениями обмена соединительной ткани. Симптомокомплекс, помимо видимых скелетных аномалий, включает целый ряд изменений со стороны других органов и систем, приводящих больного к инвалидности: нарушения зрения, сердечно-сосудистой системы, легких.
Не менее важным диагностическим признаком, от которого в значительной мере зависит прогноз продолжительности жизни больного, являются изменения сердечно-сосудистой системы.
Нервная система при синдроме Марфана не остается интактной. Большинство больных имеют признаки интеллектуальной недостаточности (IQ — не более 79 ед.), которая нередко сочетается с психопатическими и эмоционально-волевыми нарушениями, реже - с шизофреноподобными состояниями.
Для ряда больных свойственно состояние повышенной возбудимости, наличие суицидальных мыслей, быстрая истощаемость нервных процессов, отсутствие волевых установок к выполнению задания и др. Биоэлектрическая активность головного мозга больных детей отличается большим разнообразием. У детей с интеллектуальной недостаточностью ЭЭГ характеризуется отсутствием регулярного возрастного ритма, низковольтной аритмией (в диапазоне дельта-, тета- и альфа-волн).
Амплитуда варьирует от 5 до 10-30 мкв у одних детей и от 10 до 40-50 мкв - у других. Слабо выражена дифференцировка зон. Реактивность на афферентные раздражители снижена. У части детей отмечаются признаки ирритации подкорковых образований в виде диффузных веретенообразных вспышек альфа-волн, тета-ритмов, единичных рассеянных острых волн. В ЭЭГ детей с психопатическими, эмоционально-волевыми и шизофрено-подобными нарушениями на первый план выступают изменения корковой нейродинамики с нарушением правильных взаимоотношений коры и подкорковых образований.
Казалось бы, что при таком своеобразном симптомокомплексе диагноз синдрома Марфана не должен вызывать особых трудностей. Тем не менее, сложность диагностики нередко объясняется клиническим полиморфизмом, когда нет полного симптомокомплекса и болезнь ограничивается лишь несколькими симптомами:
Полная триада клинических симптомов включает:
• нарушения опорно-двигательного аппарата,
• расстройства зрения,
• нарушения сердечно-сосудистой деятельности. Неполный симптомокомплекс может быть в 2-х вариантах:
• нарушения опорно-двигательного аппарата в сочетании с расстройствами зрения и
• нарушения опорно-двигательного аппарата в сочетании с патологией сердечно-сосудистой системы.
Нарушения зрения при синдроме Марфана:
1. Подвывих и вывих хрусталиков.
2. Миопия слабой и средней степени тяжести.
3. Изменения глазного дна:
- расширение вен глазного дна,
- дегенерация сетчатой оболочки глаза,
- отслойка сетчатки.
4. Вторичная глаукома.
5. Гетерохромия радужки.
Изменения сердечно-сосудистой системы при синдроме Марфана:
- Нарушение проводящей системы сердца (по данным ЭКГ).
- Функциональные сердечные расстройства.
- Нарушение метаболических процессов в миокарде.
- Поражение митрального клапана и наличие клинически выраженного митрального порока сердца.
- Аневризма аорты и аортальные пороки (как причина внезапной смерти).
- Врожденные пороки сердца (атриовентрикулярная коммуникация).
При оценке наблюдаемых симптомов следует иметь в виду, что полный симптомокомплекс формируется постепенно. Чем старше ребенок, тем скорее можно ожидать формирование полного симптомокомплекса за счет прогредиентного течения патологического процесса.
Однако при обследовании ребенка с подозрением на синдром Марфана следует иметь в виду еще два весомых аргумента в пользу предполагаемого диагноза: тип наследования и показатели обмена соединительной ткани.

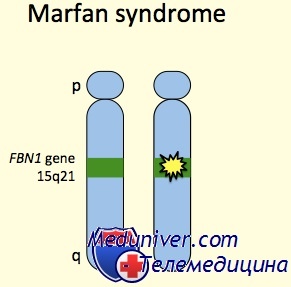
Хорошо известно, что синдром Марфана относится к наследственным заболеваниям с аутосомно-доминантным типом наследования. Более того, установлена локализация мутации - в фибриллин 1 гене (FBN1). При этом риск рождения больного ребенка достигает 50%. Рисунок демонстрирует повторное рождение детей с синдромом Марфана, мать которых страдала аналогичным заболеванием.
Окончательным аргументом в пользу диагноза синдрома Марфана является обнаружение нарушений в обмене кислых гликозаминогликанов (ГАГ) и оксипролина (ОП). При синдроме Марфана изменяются почти все фракции кислых ГАГ (хондроитинсульфаты А и С, гиалуроновая кислота, гепаран- и дерматансульфаты). Особенно высокая концентрация хондроитинсульфатов А и С обнаруживается у больных, в клинической картине которых доминировали грубые скелетные аномалии. Это объясняется тем, что хондроитинсульфат является главным гликозаминогликаном соединительной ткани.
Таким образом, окончательный диагноз синдрома Марфана ставится на основании обнаружения следующих признаков (А.Н. Семячкина):
• Нарушение опорно-двигательного аппарата.
• Нарушение зрения - подвывих хрусталика.
• Изменения сердечно-сосудистой деятельности - нарушенная проводимость, формирование аневризмы аорты.
• Аутосомно-доминантный тип наследования.
• Высокая экспрессия кислых ГАГ, особенно хондроитинсульфатов.
В последние годы в лечении синдрома Марфана достигнуты определенные успехи. Однако они определяются не целенаправленным воздействием на патогенетические механизмы этого заболевания, а на коррекцию отдельных патологических состояний:
• хирургическое лечение подвывиха хрусталика,
• хирургическую коррекцию сетчатой оболочки глаза,
• хирургическую реконструкцию аневризмы аорты.
Все это не излечивает, но позволяет значительно улучшить качество жизни больного. Основным заболеванием, с которым необходимо проводить дифференциальный диагноз, является гомоцистинурия, имеющая аналогичный фенотип.
- Рекомендуем далее ознакомиться со статьей "Гомоцистинурия - фенотип, диагностика"
Оглавление темы "Наследственные заболевания у детей":- Синдром Марфана - фенотип, диагностика
- Гомоцистинурия - фенотип, диагностика
- Гиперлипопротеидемия (ГЛП) - фенотип, диагностика
- Синдром Кнаппа-Комровера (наследственная витамин В6-зависимая ксантуренурия) - фенотип, диагностика
- Несовершенное костеобразование (НК) - фенотип, диагностика
- Врожденный множественный артрогрипоз (ВМА) - фенотип, диагностика
- Синдром Дауна - фенотип, диагностика
- Основные причины задержки психомоторного развития детей
- Основные причины микроцефалии
- Основные причины макроцефалии