
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Гомоцистинурия - фенотип, диагностика
На консультативном приеме врач обследует ребенка, который по своим внешним признакам напоминает больного с синдромом Марфана.
Фенотипический портрет больного ребенка:
I. Особенности строения скелета:
• Укороченное туловище,
• Удлиненные конечности,
• Нарушение осанки,
• «Башенная» форма черепа,
• Неправильный прикус и рост зубов,
• Высокое небо,
• Короткая шея,
• «Крыловидные» лопатки,
• Воронкообразная деформация грудной клетки (I степени),
• Вальгусная деформация нижних конечностей,
• Плоскостопие,
• Выступление пяточных бугров,
• Остеопороз (незначительно и умеренно выраженный).
II. Изменения нервной системы:
• Снижение интеллекта,
• Патологический характер ЭЭГ
• Спастическая походка.
III. Расстройства зрения:
• Подвывих хрусталиков)!)
• Вторичная глаукома,
• Изменения глазного дна (некоторое обесцвечивание сетчатки),
IV. Изменения сердечно-сосудистой системы:
• Нарушения обменных процессов в миокарде (по данным ЭКГ)
V. Светлые, мягкие волосы, вьющиеся крупными завитками.
VI. Цвет радужной оболочки глаз:
• Голубой,
• Карий.
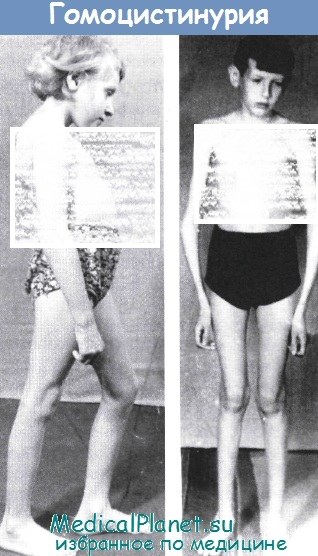
Действительно, как и при болезни Марфана, в патологический процесс вовлечены: скелет, нервная система и даже такой редкий и своеобразный симптом как подвывих хрусталика. Более того, выявляемые изменения обмена соединительной ткани весьма похожи на те же хромотограммы, которые свойственны больным с синдромом Марфана (различие лишь карбазолорциноловых коэффициентов кислых ГАГ). Однако различие между этими фенотипически схожими, но генетически гетерогенными заболеваниями, чрезвычайно велики, т.к. речь может идти о синдроме Марфана и гомоцистинурии.
• При гомоцистинурии отсутствует кардинальный для синдрома Марфана симптом - арахнодактилия (!),
• Гомоцистинурия имеет аутосомно-рецессивный тип наследования,
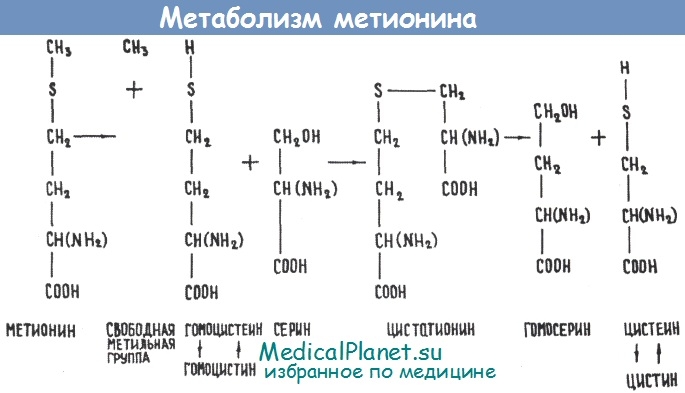
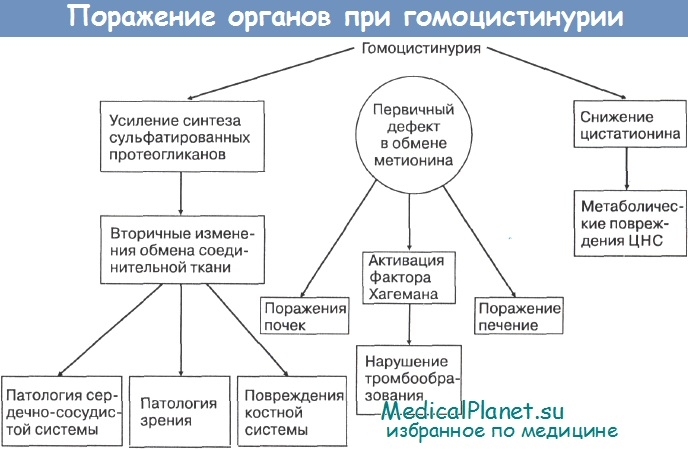
• Гомоцистинурия является наследственным заболеванием обмена аминокислоты метионина в результате врожденного дефицита фермента цистатионин-синтетазы, а соединительная ткань включается в патологический процесс вторично,
• Умственная отсталость при гомоцистинурии носит более грубый характер (IQ < 50 ед.) и расценивается как олигофрения.
Гомоцистинурия впервые была описана в 1962 г. Биохимически она проявляется повышением концентрации метионина, гомоцистина и смешанных дисульфидов гомоцистина-цистеина. Диагноз основывается на изменениях аминокислотного спектра крови и мочи: повышение концентрации метионина и обнаружение несвойственных для здоровых людей гомоцистина и смешанных дисульфидов гомоцистеина-цистеина.
Снижение уровня цистатионина оказывает повреждающее влияние на развивающуюся нервную систему, т. к. цистатионин необходим для нормального развития и деятельности головного мозга. Наряду с этим склонность к тромбозам церебральных сосудов при гомоцистинурии способствует возникновению некротически-дегенеративных изменений в мозговой ткани. Не исключено, что гомоцистеинемия способствует извращению обмена соединительной ткани, т. к. при этом обнаруживают усиление синтеза сульфатированных протеогликанов с последующей дегенерацией эластических элементов, излишней депозицией коллагена, кальцификацией и гиперплазией миоинтимальных артериальных стенок.
Очень существенно то, что при гомоцистинурии в патологический процесс вовлекается такой важный орган, как печень. В ней происходят значительные структурные и функциональные изменения: дистрофические и компенсаторно-приспособительные изменения паренхимы. Среди обнаруживаемых нарушений печени преобладает диффузная белковая зернистая и вакуольная дистрофия, расстройства внутрипеченочного кровообращения, признаки холестаза и др. Электронная микроскопия демонстрирует симптомы дезорганизации тонкой структуры цитоплазмы гепатоцитов.

Необходимо отметить, что многие обнаруживаемые изменения оказываются весьма схожими у детей с нелеченной ФКУ.
Гомоцистинурия неоднородна, она генетически гетерогенна. Существуют два варианта заболевания, пиридоксинчувствительная и пиридоксинрезистентная формы. Активность фермента цистатионинсинтетазы может быть восстановлена путем назначения больших (не принятых в фармакологии) доз пиридоксина- до 100-500 мг/сут. При норме 0,5-1,5 мг. При таком лечении уже на 4-й день обычно происходит нормализация ранее измененных биохимических показателей: снижается до нормы концентрация метионина, исчезает гомоцистин и дисульфиды. Параллельно с этим происходит изменение уровня серина и цистина.
При пиридоксинрезистентной форме гомоцистинурии использование в лечение даже больших доз витамина В6 (до 100-300 мг/сут и более) не приводит к нормализации метаболизма. Только применение специально созданных диет с ограничением метионина позволяет нормализовать обменные нарушения. Обращает на себя внимание возможность комбинации одновременного существования двух генетически детерминированных энзимопатий: мегилмалоновая ацидурия + гомоцистинурия. При этой комбинации врожденных дефектов внутриклеточного метаболизма повреждения печени приводят к нарушениям перехода кобаламина в метилкобаламин и аденозилкобаламин.
В основе этой патологии лежит снижение активности метилмалонил-СоА-мутазы и метионинсинтетазы. Как следствие метаболических расстройств у больных детей к концу 1 -го года наступаюттяжелые неврологические повреждения белого вещества головного мозга, глиоз, диффузные интракраниальные экстрацеребральные артериальные повреждения, гидроцефалия и судороги, пигментный ретинит. Весьма высок процент врожденных пороков развития. Длительное лечение IMOH cbl и карнитином достаточно хорошо корригируют биохимические отклонения.
Относительно репродуктивной функции у женщин с гомоцистинурией, обусловленной недостаточностью цистатионин-бетасинтетазы, существует мнение, что при ранней диагностике генетического дефекта и своевременно начатом лечении, увеличивается репродуктивная способность. Однако следует помнить о риске тромбоэмболии у беременных и возникновении у них преэклампсии. Другие же авторы утверждают, что беременность при гомоцистинурии протекает без осложнений.
При комбинации метилмалоновой ацидемии и гомоцистинурии возможна пренатальная диагностика путем определения дефицита карнитина.
- Рекомендуем далее ознакомиться со статьей "Гиперлипопротеидемия (ГЛП) - фенотип, диагностика"
Оглавление темы "Наследственные заболевания у детей":- Синдром Марфана - фенотип, диагностика
- Гомоцистинурия - фенотип, диагностика
- Гиперлипопротеидемия (ГЛП) - фенотип, диагностика
- Синдром Кнаппа-Комровера (наследственная витамин В6-зависимая ксантуренурия) - фенотип, диагностика
- Несовершенное костеобразование (НК) - фенотип, диагностика
- Врожденный множественный артрогрипоз (ВМА) - фенотип, диагностика
- Синдром Дауна - фенотип, диагностика
- Основные причины задержки психомоторного развития детей
- Основные причины микроцефалии
- Основные причины макроцефалии