
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Метаболические энцефалопатии грудных детей: лейкодистрофии Краббе, Пилицеуса-Мерцбахера, болезнь Фарбера
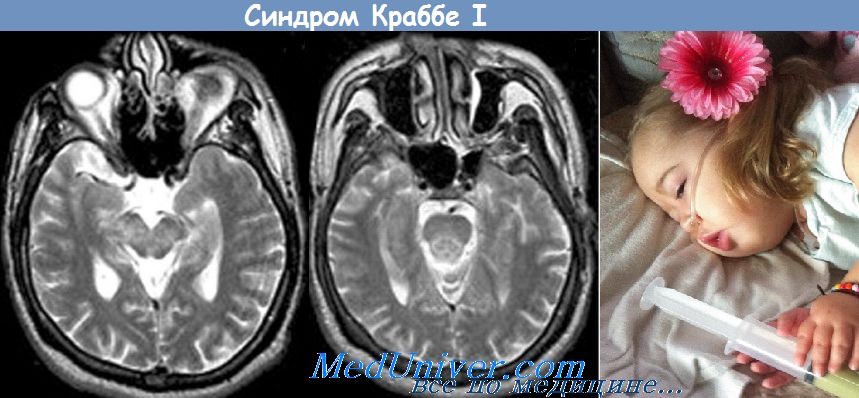
1. Лейкодистрофия Краббе (глобоидноклеточная лейкодистрофия, болезнь Краббе-Бенеке, галактозилцерамидныйлипоидоз). Манифестация ранней инфантильной формы происходит на 3-6-м месяце жизни: повышается мышечный тонус нижних конечностей, появляются плаксивость, раздражительность, постепенно формируется спастический тетрапарез, временами децеребрационная ригидность.
Глазные симптомы: спонтанный нистагм, снижение зрения, атрофия зрительных нервов, слепота. В спинномозговой жидкости повышена концентрация белка. Смерть наступает на 2-3-м году жизни. Патоморфологически: снижена масса головного мозга, расширены мозговые желудочки. Наблюдается астроцитарный глиоз. В тканях выявляется накопление галактоцереброзидов.
Тип наследования — А/Р. В основе заболевания - недостаточность галактоцереброзид-(3-галактозидазы. Дифференциальный диагноз проводится с такими непрогрессирующими энцефалопатиями, как родовая травма или пренатальное повреждение мозга (фетальный энцефалит), а также с аминоацидопатиями у новорожденных, болезнью Лея, Gm2-ганглиозидозом, хроническими болезнями желудочно-кишечного тракта и врожденными периферическими невропатиями.
2. Болезнь Фарбера (липогранулематоз). Манифестация в периоде новорожденности: узловатые эритематозные отеки запястий, в последующем изменяются и другие суставы. Неврологические нарушения складываются из пирамидных и экстрапирамидных расстройств, задержки психомоторного развития, судорог. Смерть наступает на 2-м году жизни.
Патоморфологически: липогранулемы кожи, накопление в нервной ткани церамид. Тип наследования — A/R В основе заболевания - недостаточность церамидазы. Дифференциальный диагноз проводят с нейролипидозами, особо важное значение следует придавать узловатым эритематозным отекам запястий.
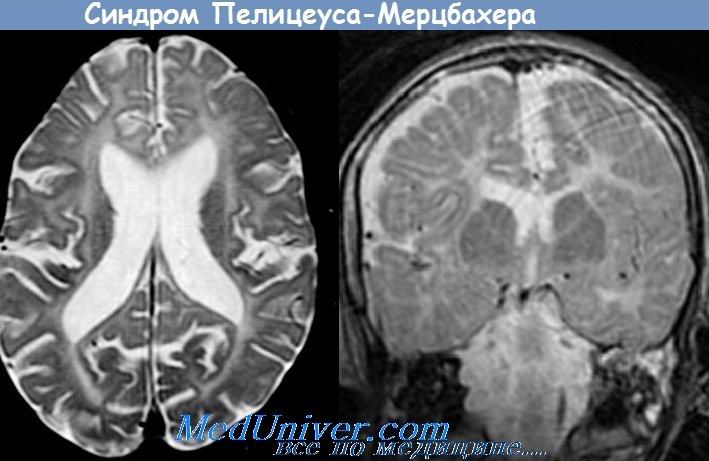
3. Лейкодистрофия Пилицеуса-Мерцбахера. Манифестация в раннем детском возрасте: спонтанный нистагм, подергивание головы и кивки. Постепенно утрачиваются двигательные навыки, нарастают спастический тетрапарез, мозжечковые симптомы. Дети умирают на 5-7-м году жизни.
Патоморфологически: диффузная демиелинизация белого вещества мозга, атрофия коры мозжечка и др. Тип наследования — рецессивный, сцепленный с Х-хромосомой. Генез заболевания неизвестен.
- Рекомендуем далее ознакомиться со статьей "Метаболические энцефалопатии грудных детей: болезни Канаван-ВанБогорта-Бертрана, Александера, Альперса"
Оглавление темы "Наследственные метаболические энцефалопатии у грудных детей":- Биохимическиая характеристика наследственных аминоацидопатий
- Метаболические энцефалопатии грудных детей: болезни Тея-Сакса и Сандхоффа
- Метаболические энцефалопатии грудных детей: болезни Гоше, Нимана-Пика
- Метаболические энцефалопатии грудных детей: лейкодистрофии Краббе, Пилицеуса-Мерцбахера, болезнь Фарбера
- Метаболические энцефалопатии грудных детей: болезни Канаван-ВанБогорта-Бертрана, Александера, Альперса
- Метаболические энцефалопатии грудных детей: болезни Лея, Менкеса, врожденный лактатацидоз
- Метаболические энцефалопатии грудных детей: болезни Целльвегера, Гирке, синдром Лоу
- Метаболические энцефалопатии грудных детей: болезнь Помпе, дефицит гликоген-синтетазы, непереносимость фруктозы
- Метаболические энцефалопатии грудных детей: болезнь Мак-Ардога, недостаточность ацил-КоА-дегидрогеназы жирных кислот со средней длиной цепи
- Дифференциация метаболических энцефалопатий грудных детей