
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Дифференциация метаболических энцефалопатий грудных детей
Сопоставляя особенности клинической картины заболеваний группы наследственных метаболических энцефалопатий грудных детей, необходимо отметить, что их фенотипическое сходство обусловлено рядом общих признаков:
• раннее начало заболевания (первые месяцы жизни);
• злокачественность течения патологического процесса (смерть наступает на 1-3 году жизни);
• тяжесть поражения многих органов и систем (ЦНС, зрения, слуха, печени, скелета и др.).
В то же время детальный анализ показывает, что, несмотря на клиническое сходство этих заболеваний, у них есть и определенные различия, которые могут быть использованы в процессе дифференциальной диагностики.
Прежде всего, заслуживают внимание особенности периода новорожденности у детей этой группы. Помимо общесоматических расстройств (анорексия, рвота, гипотрофия и др.), отмечаемых в 121 из 20 нозологических форм, при 5 заболеваниях с первых дней жизни у детей имеются бульбарные расстройства.
Следовательно, из 12 заболеваний особо могут быть выделены 5: болезнь Гоше (№4), болезнь Альперса (№11), подострый некротизирующий энцефалит (№ 12), болезнь Целльвегера (№ 15), синдром Лоу (№ 16).
Затем из этих заболеваний может быть выделен, например, синдром Лоу, при котором имеется своеобразное сочетание клинических признаков: церебральных повреждений, глазных симптомов (микрофтальмия, катаракта, глаукома, хориоретинит) и нарушений функции почек (аминоацидурия, глюкозурия, протеинурия и др.).
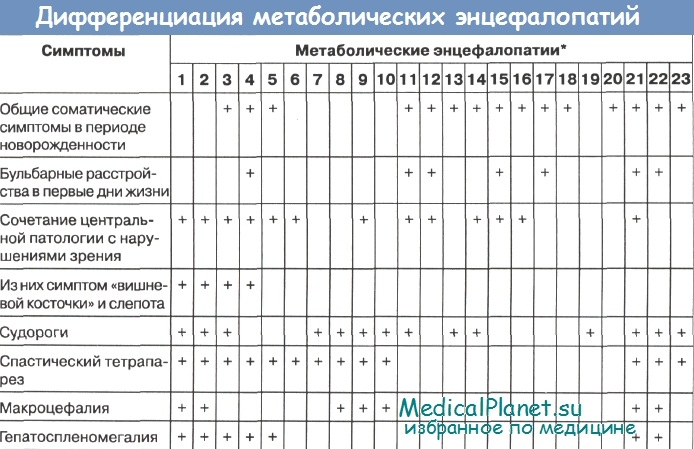
Симптомокомплекс детей в возрасте 6 месяцев и старше представлен обычно сочетанием ряда грозных симптомов. Прежде всего, это симптомы задержки психомоторного развития. Практически они ярко выражены при всех 20 заболеваниях этой группы. Церебральные нарушения особенно часто сочетаются с патологией зрения (снижение зрения, атрофия зрительного нерва, слепота и др.). Такое сочетание наблюдается при 12 заболеваниях из 20 нозологических форм.
Из этих 12 заболеваний особенно следует выделить 4, при которых задержка психомоторного развития сочетается со своеобразной патологией зрения -наличие на глазном дне «вишневой косточки»: болезнь Тея-Сакса (№ 1), болезнь Сандхоффа (№ 2), ранняя инфантильная форма Gm1-ганглиозидоза (№ 3), ранняя инфантильная форма болезни Нимана-Пика (№ 4).
Таким образом, резко сужается круг заболеваний, с которыми проводится дифференциальный диагноз. Дальнейшее разграничение может пойти по принципу сопоставления наиболее важных симптомов: сочетание двигательно-акустических реакций со слепотой, «вишневой косточкой» на глазном дне может свидетельствовать в пользу болезни Тея-Сакса или же болезни Сандхоффа; увеличение печени и селезенки в сочетании с «вишневой косточкой» на глазном дне - об инфантильной форме болезни Нимана-Пика; сочетание лицевого дисморфизма с «вишневой косточкой» на глазном дне и гепатомегалией - о ганглиозидозе Gm1
Аналогичный принцип дифференциальной диагностики может быть использован и при разграничении других заболеваний, при которых церебральная патология сочетается с другими нарушениями зрения. Так, сочетание офтальмоплегии с ретракцией головы, увеличением селезенки и отсутствием на глазном дне «вишневой косточки» может свидетельствовать в пользу болезни Гоше; ранняя слепота в сочетании с атрофией зрительных нервов, тоническим спазмом разгибателей и мегалэнцефалией, а также отсутствие гепатоспленомегалии - в пользу предположительного диагноза спонгиозной дегенерации; раннее увеличение окружности головы и отсутствие нарушений зрения в сочетании с висцеромегалией - в пользу лейкодистрофии Александера; эпизодические нарушения дыхания, паралич глаз при нормальном зрении, висцеромегалия - в пользу подострой некротизирующей энцефалопатии Лея; дисморфизм лица, увеличение печени, врожденные аномалии глаз и ушей -в пользу болезни Целльвегера; сочетание церебральных нарушений с узловатыми эритематозными отеками запястий и других суставов - в пользу болезни Фарбера; сочетание церебральных и респираторных расстройств с потерей кожной пигментации и изменением волос - в пользу болезни курчавых волос.
Предлагаемые критерии дифференциальной диагностики являются сугубо ориентировочными и позволяют вести более целенаправленное обследование больного. Окончательный же диагноз основывается на комплексе клинических и лабораторных данных.
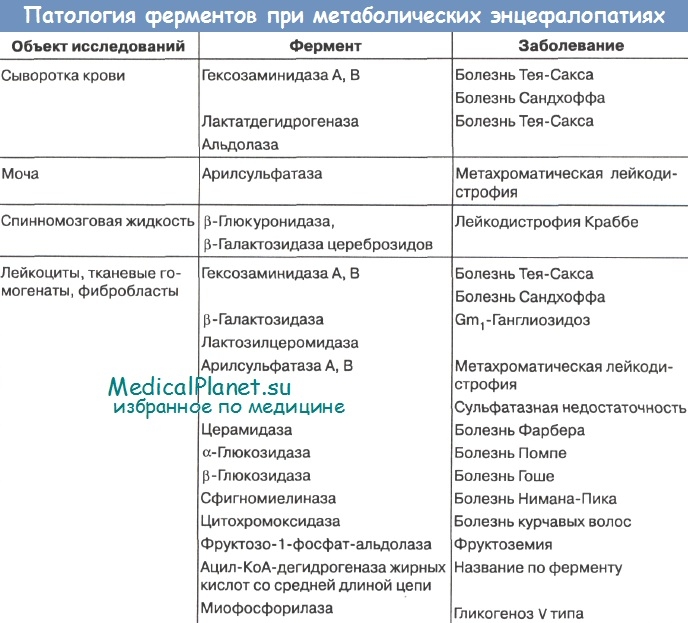
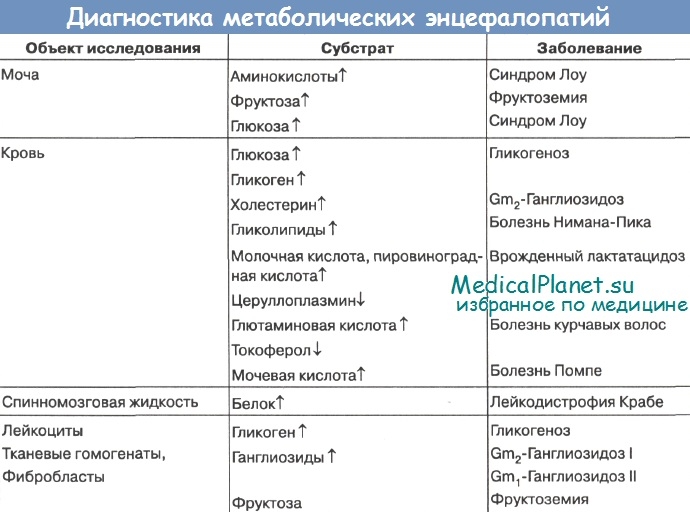
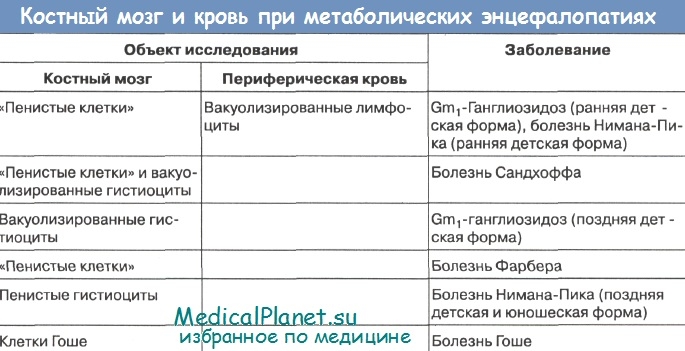
Из 20 заболеваний этой группы 6 - лейкодистрофия Пелицеуса-Мерцбахера, болезнь Канавана-Ван-Богарда-Бертранда, болезнь Александера, болезнь Альперса, болезнь Лея, болезнь Целльвегера -диагностируются только при аутопсиях по характеру патоморфологических изменений мозга и других органов. Это обусловлено тем, что до сих пор патогенетические механизмы названных страданий неизвестны.
При дифференциальной диагностике остальных 17 заболеваний могут быть использованы биохимические и морфологические методы. Наиболее достоверны при окончательной постановке диагноза методы ферментативной диагностики. Однако они доступны лишь для некоторых научно-исследовательских учреждений.
- Вернуться в оглавление раздела "генетика"
Оглавление темы "Наследственные метаболические энцефалопатии у грудных детей":- Биохимическиая характеристика наследственных аминоацидопатий
- Метаболические энцефалопатии грудных детей: болезни Тея-Сакса и Сандхоффа
- Метаболические энцефалопатии грудных детей: болезни Гоше, Нимана-Пика
- Метаболические энцефалопатии грудных детей: лейкодистрофии Краббе, Пилицеуса-Мерцбахера, болезнь Фарбера
- Метаболические энцефалопатии грудных детей: болезни Канаван-ВанБогорта-Бертрана, Александера, Альперса
- Метаболические энцефалопатии грудных детей: болезни Лея, Менкеса, врожденный лактатацидоз
- Метаболические энцефалопатии грудных детей: болезни Целльвегера, Гирке, синдром Лоу
- Метаболические энцефалопатии грудных детей: болезнь Помпе, дефицит гликоген-синтетазы, непереносимость фруктозы
- Метаболические энцефалопатии грудных детей: болезнь Мак-Ардога, недостаточность ацил-КоА-дегидрогеназы жирных кислот со средней длиной цепи
- Дифференциация метаболических энцефалопатий грудных детей