
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизмы развития (патогенез) бета-талассемии
Синдромы талассемии составляют гетерогенную группу расстройств, вызываемых наследуемыми мутациями, снижающими синтез HbА. Две a-цепи в молекуле HbА кодируются идентичной парой генов а-глобина на 16-й хромосоме, тогда как две b-цепи кодируются одиночным геном b-глобина на 11-й хромосоме.
b-Талассемия обусловлена недостаточным синтезом b-цепей, и а-талассемия — недостаточным синтезом a-цепей. Гематологические последствия сниженного синтеза одной глобиновой цепи обусловлены не только дефицитом гемоглобина, но еще и относительным избытком другой глобиновой цепи, особенно при b-талассемии.
Синдромы талассемии эндемичны в странах Средиземноморского бассейна, на Среднем Востоке и Индийском субконтиненте, в тропической Африке и Азии и являются наиболее распространенными наследственными заболеваниями. Как и в случае серповидно-клеточной анемии и других наследственных нарушений с поражением эритроцитов, распространение талассемий обусловлено тем, что гетерозиготность создает защиту против малярии.
Хотя мы рассматриваем синдромы талассемии вместе с другими наследственными анемиями, ассоциированными с гемолизом, важно учитывать, что нарушения синтеза цепей глобина, лежащие в основе талассемий, также влияют на продукцию эритроцитов и вносят свой вклад в патогенез этих заболеваний.
b-Талассемии возникают в результате мутаций, снижающих синтез b-цепей. Тяжесть заболевания варьирует из-за гетерогенности мутаций. Сначала опишем молекулярные повреждения при b-талассемии, а затем — клинические варианты, соответствующие специфическим молекулярным дефектам.
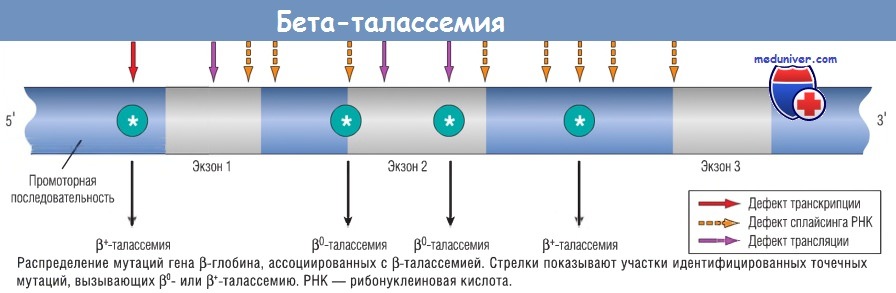
а) Молекулярный патогенез. Мутации, вызывающие b-талассемии, делят на две категории:
(1) мутации b0, ассоциированные с отсутствием синтеза b-цепей;
(2) мутации b+, характеризующиеся сниженным (но определимым) синтезом b-цепей.
Секвенирование генов b-талассемии показало, что существует более 100 различных каузальных мутаций, в основном точечных. Детальное описание этих мутаций и их эффекты изложены в специальной литературе. Приведем некоторые примеры мутаций:
- сплайсинг-мутации. Эти мутации — наиболее частая причина b+-талассемии. Большинство мутаций локализуются в интронах, и только немногие — в экзонах. Некоторые из этих мутаций нарушают нормальный сплайсинг РНК и подавляют образование нормальной b-глобиновой мРНК, в результате развивается b0-талассемия. Другие мутации создают в интроне эктопический участок сплайсинга. Поскольку остаются фланкированные нормальные участки сплайсинга, происходит как нормальный, так и аномальный сплайсинг и образуется некоторое количество нормальной b-глобиновой мРНК, что приводит к развитию b+-талассемии;
- мутации в области промотора. Эти мутации уменьшают транскрипцию на 75-80%, но синтезируется некоторое количество нормальных b-цепей. Эти мутации ассоциируются с b'-талассемией;
- мутации-терминаторы цепи. Эти мутации являются наиболее частой причиной b0-талассемии. В этой категории выделяют два типа мутаций. Наиболее частый тип приводит к образованию в экзоне нового стоп-кодона, при другом типе мутаций происходят небольшие вставки или делеции, что вызывает сдвиг рамки считывания мРНК (мутации со сдвигом рамки считывания). Мутации обоих типов блокируют трансляцию и предотвращают синтез любых функциональных b-цепей.
Нарушение синтеза b-цепей приводит к развитию анемии посредством двух механизмов: неэффективного эритропоэза и внутрисосудистого гемолиза. Недостаточный синтез HbА обусловливает образование недогемоглобинизированных микроцитарных гипохромных эритроцитов со сниженной способностью к транспорту кислорода. Еще большее значение имеет уменьшение выживаемости эритроцитов и их клеток-предшественников, возникающее в результате дисбаланса синтеза а- и b-цепей.
Неспаренные а-цепи преципитируют внутри клеток-предшественников эритроцитов, образуя нерастворимые включения, которые вызывают ряд нежелательных эффектов. Однако важнейшая причина патологии эритроцитов — повреждение мембраны. Многие клетки-предшественники эритроцитов с поврежденной мембраной подвергаются апоптозу. При тяжелой b-талассемии 70-85% таких клеток-предшественников эритроцитов погибают в костном мозге, вследствие чего происходит неэффективный эритропоэз.
Эритроциты, вышедшие из костного мозга, также имеют включения и повреждения мембраны, в связи с чем склонны к секвестрации в селезенке и внесосудистому гемолизу.
При тяжелой b-талассемии неэффективный эритропоэз создает ряд дополнительных проблем. Эритропоэз в условиях тяжелой некомпенсированной анемии приводит к массивной эритроидной гиперплазии в костном мозге и интенсивному экстрамедуллярному эритропоэзу. Клетки-предшественники эритроцитов разрушают кортикальный слой кости и нарушают рост костей, что приводит к аномалиям скелета.
Экстрамедуллярный гемопоэз происходит в печени, селезенке и лимфоузлах, а в тяжелых случаях внекостные массы гемопоэтической ткани образуются в грудной, брюшной полостях и полости таза. В отсутствие лечения метаболически активные эритроидные клетки-предшественники лишают питательных веществ ткани, уже страдающие от недостатка кислорода, вызывая тяжелую кахексию.
Другое серьезное осложнение неэффективного эритропоэза — избыточная абсорбция содержащегося в пище железа. При неэффективном эритропоэзе снижается уровень циркулирующего гепсидина, важнейшего отрицательного регулятора абсорбции железа. Низкий уровень гепсидина и поступление железа при повторных гемотрансфузиях неизбежно приведут к переизбытку железа в организме, если не принять предупредительных мер. Часто происходит вторичное поражение паренхиматозных органов, особенно перегруженной железом печени; иногда развивается вторичный гемохроматоз.
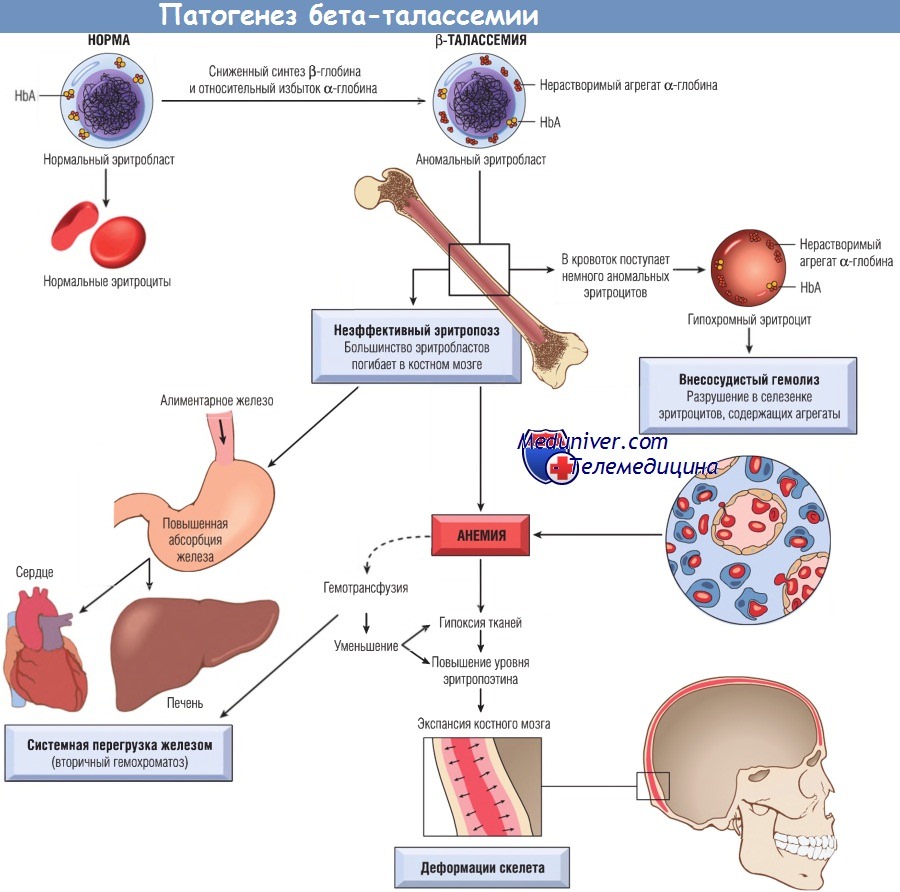
Обратите внимание, что агрегаты неспаренных цепей b-глобина (отличительный признак заболевания) не видны в мазках крови, окрашенных обычным методом.
Гемотрансфузия представляет собой обоюдоострое оружие: она уменьшает анемию и число сопутствующих осложнений, однако способствует системной перегрузке организма железом.
Hb — гемоглобин.
б) Клинические синдромы. Соотношение между клиническими синдромами и определяющими их генотипами представлено в таблице ниже. Клиническая классификация b-талассемий основана на тяжести анемии, которая, в свою очередь, зависит от генетического дефекта (b+ или b0) и дозы гена (гомо- или гетерозиготность).
Индивиды с двумя аллелями b-талассемии (b+/b+, b+/b0 или b0/b0) имеют тяжелую, трансфузионно-зависимую анемию — большую b-талассемию. Гетерозиготы с одним геном b-талассемии и одним нормальным геном (b+/b или b0/b) обычно имеют слабую бессимптомную микроцитарную анемию. Это состояние называют малой b-талассемией или признаком b-талассемии. Третью категорию — генетически гетерогенную и умеренной тяжести — называют промежуточной b-талассемией.
Эта категория включает более легкие варианты b+/b+ - или b+/b0-талассемии и необычные формы гетерозиготной b-талассемии. Некоторые пациенты с промежуточной b-талассемией имеют два дефектных гена b-глобина и дефект гена а-талассемии, который уменьшает дисбаланс между синтезом а- и b-цепей. В других редких случаях присутствуют одиночный дефект b-глобина и 1-2 экстракопии нормальных генов а-глобина (результат дупликации гена), усиливающие дисбаланс цепей. Эти необычные формы заболевания подчеркивают кардинальную роль неспаренных цепей а-глобина в развитии данной патологии.
Клинические и морфологические признаки промежуточной b-талассемии не будут рассмотрены отдельно, поскольку представление о них можно получить после описания большой b-талассемии и малой b-талассемии.
- Большая b-талассемия. Наиболее распространена в странах Средиземноморского бассейна, некоторых районах Африки и в Юго-Восточной Азии. В США заболевание чаще всего регистрируют у иммигрантов из этих регионов. Анемия проявляется через 6-9 мес после рождения, по мере того как происходит переключение синтеза гемоглобина с HbF на НЬА. В отсутствие гемотрансфузии уровень гемоглобина у пациентов составляет 3-6 г/дл. HbА в эритроцитах полностью отсутствует (генотип b0/b0) или содержится в небольшом количестве (генотипы b+/b+ или b+/b0).
Основной гемоглобин в эритроцитах — HbF, уровень которого заметно повышен. Содержание HbА2 иногда увеличено, но чаще является нормальным или низким.

Морфология. В мазках крови видны выраженные аномалии эритроцитов, включая заметные различия по величине (анизоцитоз) и форме (пойкилоцитоз), а также микроцитоз и гипохромию. Обычным является также присутствие клеток-мишеней, базофильной зернистости и фрагментированных эритроцитов. Агрегаты а-глобина эффективно удаляются в селезенке, и обнаружить их нелегко. Количество ретикулоцитов повышено, но ниже ожидаемого (если судить по тяжести анемии) вследствие неэффективного эритропоэза.
В периферической крови присутствует вариабельное количество ядросодержащих клеток-предшественников эритроцитов (нормобластов) с малым количеством гемоглобина. Наличие этих клеток обусловлено «стрессом» эритропоэза и аномальным высвобождением эритроцитов из очагов экстрамедуллярного гемопоэза.
Другие значимые изменения затрагивают костный мозг и селезенку. В отсутствие гемотрансфузии у пациента происходит резко выраженная экспансия гемопоэтически активного костного мозга. В костях лица и черепа растущая костномозговая ткань разрушает существующий кортикальный слой кости и индуцирует образование новой кости. На рентгенограмме это напоминает стрижку волос «ежиком». И гиперплазия фагоцитов, и экстрамедуллярный гемопоэз способствуют увеличению селезенки, масса которой может достигать 1500 г. Печень и лимфоузлы также увеличиваются вследствие экстрамедуллярного гемопоэза.
Гемосидероз и вторичный гемохроматоз — два проявления избыточного количества железа в организме — регистрируют почти у всех пациентов. Отложения железа часто повреждают органы, в наибольшей степени сердце, печень и поджелудочную железу.
Естественное течение большой b-талассемии кратковременно. Дети имеют задержку роста и умирают в раннем возрасте от анемии, если им не проводят гемотрансфузию. Если дети с большой b-талассемией живут несколько лет, у них увеличиваются и деформируются скуловые кости и другие костные выступы. Обычно наблюдается гепатоспленомегалия вследствие экстрамедуллярного гемопоэза. Гемотрансфузии облегчают течение анемии и тормозят развитие осложнений, связанных с избыточным гемопоэзом, но сами могут стать причиной осложнений.
Болезнь сердца, развивающаяся в результате прогрессирующей перегрузки железом и вторичного гемохроматоза, нередко приводит к летальному исходу, особенно среди пациентов с многочисленными гемотрансфузиями, которых необходимо лечить комплексонами, связывающими железо, чтобы предотвратить вторичный гемохроматоз или уменьшить его проявления. Гемотрансфузии и комплексоны дают возможность пациентам дожить до 20 лет, однако общий прогноз остается неутешительным. Единственным методом, позволяющим рассчитывать на излечение, служит трансплантация костного мозга, и ее проводят все чаще. Возможна пренатальная диагностика с помощью молекулярного анализа ДНК.
- Малая b-талассемия. Встречается значительно чаще, чем большая b-талассемия, и распространена в тех же этнических группах. Большинство пациентов являются гетерозиготными носителями аллеля b+ или b0. Симптомы заболевания обычно отсутствуют или выражены слабо. В мазках периферической крови типично наличие некоторых аномалий эритроцитов, включая микроцитоз, гипохромию, базофильную зернистость и клетки-мишени. В костном мозге обнаруживается слабая эритроидная гиперплазия. При электрофорезе гемоглобина обычно обнаруживают увеличение содержания HbА2 до 4-8% общего уровня (в норме 2,5 ± 0,3%), что отражает повышенную разницу между синтезом δ-цепи и b-цепи. Уровень HbF обычно нормальный, иногда слабо повышен.
Диагностика малой b-талассемии важна по двум причинам:
(1) чтобы отличить ее от микроцитарной гипохромной железодефицитной анемии;
(2) для генетического консультирования.
Дефицит железа обычно удается исключить после оценки содержания железа и уровня ферритина в сыворотке и общей железосвязывающей способности. Определение уровня НЬА2 диагностически важно, особенно у лиц, входящих в группу риска развития как малой b-талассемии, так и дефицита железа (например, у женщин в репродуктивном возрасте).
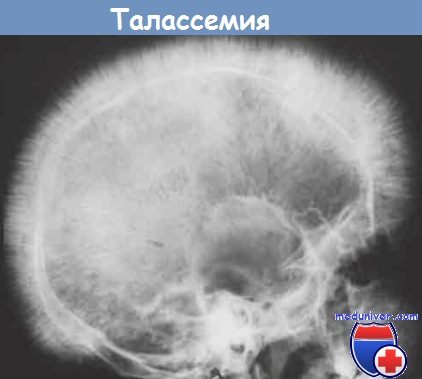
Перпендикулярные радиальные выросты напоминают стрижку волос «ежиком».
- Рекомендуем ознакомиться со следующей статьей "Механизмы развития (патогенез) альфа-талассемии"
Оглавление темы "Патогенез анемий":- Механизмы развития (патогенез) серповидно-клеточной анемии
- Механизмы развития (патогенез) бета-талассемии
- Механизмы развития (патогенез) альфа-талассемии
- Механизмы развития (патогенез) пароксизмальной ночной гемоглобинурии
- Механизмы развития (патогенез) иммуногемолитической анемии
- Механизмы развития (патогенез) мегалобластной анемии
- Механизмы развития (патогенез) пернициозной анемии
- Механизмы развития (патогенез) анемии при дефиците фолиевой кислоты
- Механизмы развития (патогенез) железодефицитной анемии
- Механизмы развития (патогенез) анемии из-за хронического заболевания