
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизмы развития (патогенез) серповидно-клеточной анемии
Серповидно-клеточная анемия представляет собой довольно частую наследственную гемоглобинопатию, возникающую в основном у лиц с темным цветом кожи. Известно несколько сотен различных гемоглобинопатий, обусловленных мутациями генов глобина, но заслуживают обсуждения лишь те, которые ассоциируются с серповидно-клеточной анемией. Напомним, что гемоглобин (Hb) является тетрамерным белком, состоящим из двух пар цепей глобина, каждая из которых имеет свою группу гема.
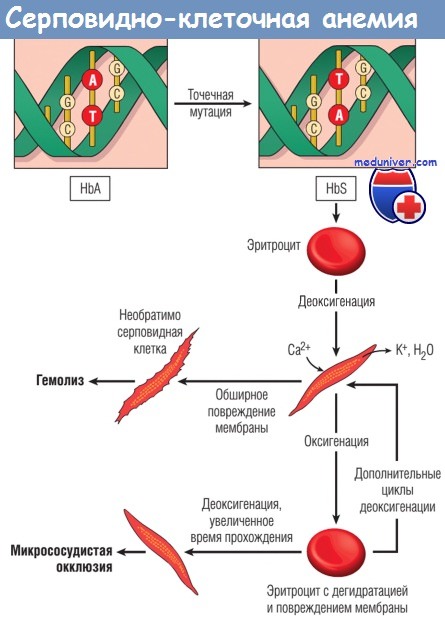
Нормальные зрелые эритроциты содержат главным образом гемоглобин А (HbA; а2b2), а также некоторое количество гемоглобина А2 (HbА2; a2δ2) и фетального гемоглобина (HbF; а2γ2). Серповидно-клеточная анемия возникает в результате точечной мутации в кодоне 6 гена b-глобина, приводящей к замене остатка глутамата остатком валина. За развитие заболевания ответственны измененные физико-химические свойства образующегося гемоглобина S (HbS; a2bs2).
Около 8-10% лиц с темным цветом кожи гетерозиготны по HbS (в США 2 млн человек). Это в основном бессимптомное состояние, известное как признак серповидно-клеточности. Дети двух гетерозигот имеют шанс 1 : 4 родиться гомозиготами по данной мутации, т.е. с симптоматической серповидно-клеточной анемией. У таких индивидов почти весь гемоглобин в эритроцитах является HbS. В США насчитывают 70 тыс. лиц с серповидно-клеточной анемией. В некоторых популяциях в Африке число гетерозигот достигает 30%. Вероятно, столь высокие цифры являются следствием защиты, создаваемой HbS от малярии, вызываемой Р. falciparum.
а) Патогенез. Молекулы HbS полимеризуются при деоксигенации. По мере образования агрегатов HbS цитозоль эритроцитов сначала превращается из свободно текущей жидкости в вязкий гель. При продолжающейся деоксигенации агрегаты HbS формируют в эритроцитах длинные, похожие на иглы волокна, вследствие чего образуются серповидные или похожие на листья падуба (остролиста) эритроциты.
Присутствие HbS лежит в основе главных патологических проявлений серповидно-клеточной анемии:
(1) хронического гемолиза;
(2) окклюзии микрососудов;
(3) повреждения тканей.
На скорость развития и тяжесть заболевания влияют различные факторы:
- взаимодействие HbS с другими типами гемоглобина в эритроците. У гетерозигот с признаком серповидно-клеточности « 40% гемоглобина представлено HbS, а оставшаяся часть — HbA, который препятствует полимеризации HbS. В результате эритроциты у гетерозигот не являются серповидными, исключая состояние глубокой гипоксии. HbF ингибирует полимеризацию HbS в еще большей степени, чем HbА, поэтому у детей симптомы не наблюдаются до возраста 5-6 мес, когда наступает естественное падение уровня HbF. Однако у некоторых индивидов уровень экспрессии HbF остается достаточно высоким — состояние, известное как наследственная персистенция HbF.
У таких пациентов серповидно-клеточная анемия гораздо менее тяжелая. Существует и другой вариант гемоглобина — HbС, у которого лизин замещен глутамином в позиции 6 аминокислотной последовательности гена b-глобина. В эритроцитах с HbSC содержание HbS составляет 50% по сравнению с 40% HbS в эритроцитах с HbAS. Кроме того, эритроциты с HbSC имеют тенденцию утрачивать соли и воду и становиться дегидратированными, что повышает внутриклеточную концентрацию HbS. Оба эти фактора усиливают тенденцию к полимеризации HbS. Вследствие этого у лиц с HbSC развивается симптоматическое серповидно-клеточное расстройство (гемоглобинопатия HbSC), менее тяжелое, чем серповидно-клеточная анемия. Примерно 1 из 1250 человек имеет гемоглобинопатию HbSC. Около 2-3% лиц с темным цветом кожи гетерозиготны по HbС и не имеют симптомов;
- средняя концентрация гемоглобина в эритроците. Более высокие концентрации HbS повышают вероятность агрегации и полимеризации во время деоксигенации. Так, дегидратация, увеличивающая среднюю концентрацию гемоглобина в эритроците, облегчает образование серповидных эритроцитов. И наоборот, условия, снижающие среднюю концентрацию гемоглобина в эритроците, уменьшают тяжесть заболевания. Это происходит, когда индивид гомозиготен по HbS, но также одновременно присутствует а-талассемия, снижающая синтез Hb, что приводит к более мягкому течению серповидно-клеточной анемии;
- внутриклеточный pH. Снижение pH уменьшает аффинность гемоглобина к кислороду, тем самым увеличивая фракцию деоксигенированного HbS при любом уровне напряжения кислорода и усиливая тенденцию к образованию серповидных форм;
- время перехода эритроцитов по микрососудам. Как будет указано далее, значительная часть патологических явлений при серповидно-клеточной анемии обусловлена микрососудистой окклюзией, вызванной серповидными эритроцитами. Время перемещения клеток в нормальных микрососудах слишком мало для значительной агрегации деоксигенированного HbS, поэтому образование серповидных форм идет там, где переход совершается медленно, — в нормальной селезенке и костном мозге (которые в результате этого существенно повреждаются при серповидно-клеточной анемии), а также в сосудистом ложе на фоне воспаления. Ток крови через воспаленные ткани замедляется вследствие адгезии эритроцитов и лейкоцитов к активированным эндотелиальным клеткам и выхода жидкости через сосуды с повышенной проницаемостью в результате воспаления. Вследствие этого сосудистое ложе склонно к формированию серповидных форм и окклюзии. Серповидные эритроциты могут усиленно экспрессировать некоторые молекулы адгезии, участвующие в связывании с эндотелиальными клетками.
Есть также данные о том, что серповидные эритроциты способны в определенной степени активировать эндотелий, что может содействовать адгезии эритроцитов и гранулоцитов, гипоксии, индуцированной окклюзией сосудов, и другим изменениям.
Образование серповидных форм обусловлено кумулятивным повреждением эритроцитов разными механизмами. По мере усиления полимеризации HbS выпячиваются через скелет мембраны из клетки, покрытой только липидным бислоем. Это серьезное нарушение скелета мембраны вызывает приток Са2+, индуцирующих перекрестное связывание мембранных белков и активирующих ионные каналы, через которые происходит отток К+ и воды. В случае повторных эпизодов болезни эритроциты становятся все более дегидратированными, плотными и ригидными. В итоге наиболее поврежденные клетки превращаются в необратимо серповидные эритроциты (сохраняющие серповидную форму даже при полной оксигенации). Тяжесть гемолиза коррелирует с процентом необратимо серповидных эритроцитов, которые быстро секвестрируются и удаляются мононуклеарными фагоцитами (внесосудистый гемолиз). Серповидные эритроциты также нестойки при механических воздействиях, что приводит к внутрисосудистому гемолизу определенной степени.
Патогенез микрососудистой окклюзии, ответственной за наиболее серьезные клинические проявления, менее изучен. Микрососудистая окклюзия не связана с количеством необратимо серповидных эритроцитов в крови, а может зависеть от ультраструктурных повреждений мембраны эритроцитов и других факторов, в частности воспаления, замедляющего или останавливающего перемещение эритроцитов через микрососудистое ложе. Как указано ранее, серповидные эритроциты экспрессируют более высокий, чем в норме, уровень молекул адгезии и обладают адгезивными свойствами. Медиаторы, высвобождаемые гранулоцитами в ходе воспалительной реакции, повышают экспрессию молекул адгезии эндотелиальными клетками и еще больше усиливают тенденцию эритроцитов задерживаться во время перехода по микрососудам. О возможной роли воспалительных клеток свидетельствует тот факт, что количество лейкоцитов коррелирует с частотой вазоокклюзионных кризов и других проявлений повреждений тканей. Застой эритроцитов в воспаленном сосудистом ложе приводит к длительному снижению напряжения кислорода, образованию серповидных форм и микрососудистой окклюзии.
Начинается цепь событий, создающих порочный круг: образование серповидных эритроцитов, обструкция, гипоксия и дальнейшее формирование серповидных эритроцитов.
Снижение уровня оксида азота (NO) также играет определенную роль в сосудистой окклюзии. Свободный гемоглобин, высвобождаемый из лизированных серповидных эритроцитов, может связывать и инактивировать NO, обладающий свойствами сильного вазодилататора и ингибитора агрегации тромбоцитов. Снижение уровня NO повышает сосудистый тонус (происходит сужение сосудов) и усиливает агрегацию тромбоцитов. Оба эффекта способствуют стазу, формированию серповидных эритроцитов и тромбозу (в некоторых случаях).
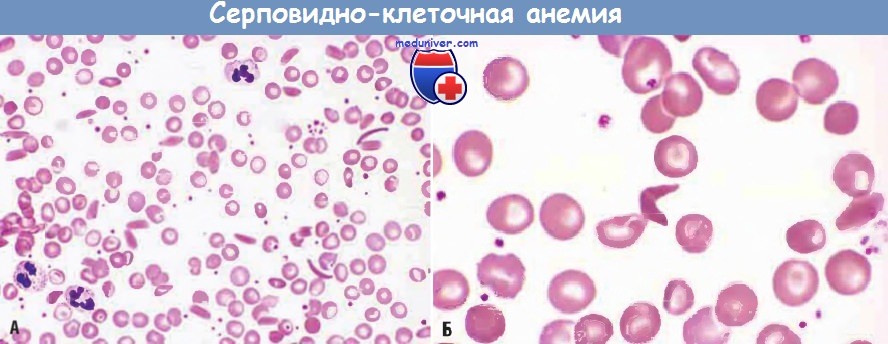
(А) При малом увеличении видны серповидные клетки, анизоцитоз и пойкилоцитоз.
(Б) При большом увеличении можно видеть в центре необратимо серповидную клетку.
б) Морфология. При полностью развившейся серповидно-клеточной анемии в периферической крови обнаруживаются значительное количество серповидных клеток, ретикулоцитоз и клетки-мишени (названные так потому, что гемоглобин скапливается в центре клеток), образующиеся в результате дегидратации эритроцитов. В некоторых эритроцитах также присутствуют тельца Хауэлла-Жолли (мелкие темные остатки ядра) вследствие асплении. Костный мозг гиперпластичный в результате компенсаторной эритроидной гиперплазии. Экспансия костного мозга приводит к костной резорбции и вторичному остеогенезу, что проявляется деформациями костей черепа. Также возможен экстрамедуллярный гемопоэз. Повышенное расщепление гемоглобина способно вызвать образование пигментных желчных камней и гипербилирубинемию.
В раннем детстве селезенка увеличивается до 500 г вследствие застоя крови в красной пульпе, обусловленного накоплением серповидных эритроцитов в тяжах и синусоидах. Однако со временем хронический стаз эритроцитов приводит к инфарктам селезенки, фиброзу и прогрессирующему сморщиванию органа, и уже в подростковом возрасте или у молодых взрослых от селезенки остается лишь незначительное количество фиброзной ткани. Этот процесс носит название аутоспленэктомии. Инфаркты, обусловленные сосудистыми окклюзиями, могут происходит во многих других тканях, включая кости, головной мозг, почки, печень, сетчатку глаза и сосуды легких (в последнем случае иногда развивается легочное сердце). У взрослых пациентов застой крови в сосудах подкожно-жировой ткани часто приводит к образованию язв нижних конечностей, у детей это осложнение наблюдается очень редко.
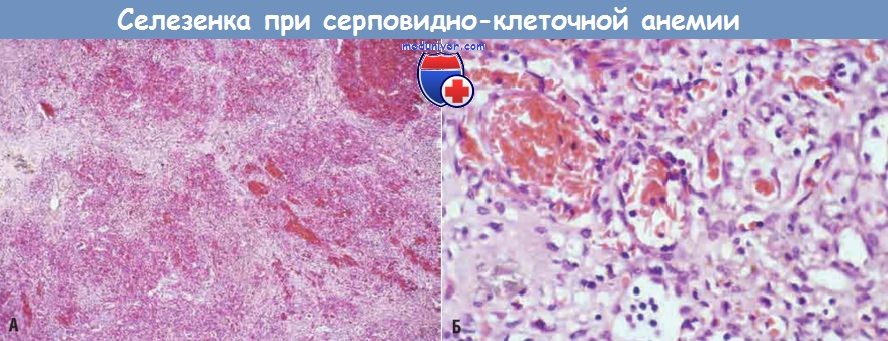
Тяжи красной пульпы и синусоиды заметно застойны; между районами застоя видны светлые участки фиброза, возникшего в результате ишемического повреждения.
(Б) При большом увеличении видны расширенные селезеночные синусоиды, заполненные серповидными эритроцитами.
в) Клинические признаки. Серповидно-клеточная анемия вызывает умеренно тяжелую гемолитическую анемию (уровень гематокрита 18-30%), ассоциированную с ретикулоцитозом, гипербилирубинемией и присутствием серповидных эритроцитов. Течение заболевания прерывается разнообразными вазоокклюзионными кризами {болевыми кризами), которые представляют собой эпизоды гипоксического повреждения и некроза, вызывающие сильную боль в пораженной области. Пусковым стимулом могут быть инфекция, дегидратация и ацидоз (все эти факторы способствуют образованию серповидных эритроцитов), однако в большинстве случаев причину установить не удается. Объектом поражения в основном служат кости, легкие, печень, головной мозг, селезенка и пенис. У детей очень часто отмечаются костные вазоокклюзионные кризы, которые нередко трудно отличить от проявлений острого остеомиелита. Поражение часто проявляется в виде синдрома «кисть-стопа» (дактилит костей стоп и/или кистей). Острый грудной синдром — особенно опасный тип вазоокклюзионного криза, поражающего легкие. Обычно отмечаются лихорадка, кашель и легочные инфильтраты.
Воспаление замедляет кровоток в ткани легких, что приводит к образованию серповидных эритроцитов и сосудистой окклюзии. Функция легких нарушается, и создается потенциально фатальный порочный круг: усиление легочной и системной гипоксемии ведет к образованию серповидных эритроцитов, что, в свою очередь, усиливает гипоксемию. Другие формы сосудистой окклюзии, особенно инсульт, также могут стать жизнеугрожающими состояниями. Предрасполагающими факторами служат адгезия серповидных эритроцитов к эндотелию артерий и вазоконстрикция, обусловленная связыванием NO свободным гемоглобином. Вазоокклюзионные кризы представляют собой наиболее частую причину смерти пациентов, но течение заболевания осложняют и другие острые процессы.
Секвестрационные кризы наблюдаются у детей с интактной селезенкой. Значительное накопление серповидных эритроцитов приводит к быстрому увеличению селезенки, гиповолемии, иногда к шоку. В некоторых случаях эти осложнения могут привести к летальному исходу. При секвестрационном кризе и остром грудном синдроме необходима экстренная обменная гемотрансфузия.
Апластические кризы происходят в результате инфицирования клеток-предшественников эритроцитов парвовирусом В19, вызывающим временное прекращение эритропоэза, в результате которого анемия утяжеляется.
Помимо неблагоприятного воздействия кризов отрицательное влияние, хотя и не сразу замечаемое, оказывает хроническая системная гипоксия. Она ответственна за нарушение роста и развития организма, а также повреждение органов, включая селезенку, сердце, почки и легкие. Образование серповидных эритроцитов на фоне повышенного давления в мозговом веществе почек вызывает повреждения, приводящие со временем к гипостенурии (неспособности концентрировать мочу), которая создает условия для дегидратации с присущим ей риском.
Другой опасностью является повышенная восприимчивость к инфекциям, вызываемым капсульными микроорганизмами. В значительной степени это связано с изменением функций селезенки в результате застоя и замедления кровотока (у детей) или инфарктов (у взрослых). Дефекты неизвестной этиологии альтернативного пути активации системы комплемента также нарушают процесс опсонизации бактерий. Частоту септицемии и менингита, вызываемых Р. pneumoniae и Н. influenzae и являющихся частой причиной смерти (особенно детей), можно снизить с помощью вакцинации и профилактического применения антибиотиков.
Следует подчеркнуть, что существуют различные клинические проявления серповидно-клеточной анемии. Некоторые пациенты страдают от повторных вазоокклюзионных кризов, тогда как у других отмечаются лишь незначительные симптомы. Причины такой широкой вариабельности заболевания неизвестны.
Диагноз ставят на основании клинических признаков и присутствия необратимо серповидных эритроцитов. Подтверждают диагноз различными тестами, с помощью которых выявляют HbS. Образцы крови смешивают с реагентами, потребляющими кислород (например, метабисульфитом), которые в случае присутствия HbS индуцируют образование серповидных эритроцитов. Используют также метод электрофореза, чтобы установить присутствие HbS и исключить серповидно-клеточные гемоглобинопатии, например гемоглобинопатию HbSC. Возможна пренатальная диагностика с помощью анализа фетальной ДНК, получаемой путем амниоцентеза или биопсии хориона.
Прогноз для пациентов с серповидно-клеточной анемией в последние 10-20 лет стал значительно лучше. Около 90% пациентов доживают до 20 лет, а 50% живут дольше 40 лет. Терапия заключается в назначении гидроксимочевины, ингибитора синтеза ДНК. Благоприятные эффекты гидроксимочевины: (1) повышение уровня HbF (механизм неизвестен); (2) противовоспалительный эффект вследствие ингибирования продукции лейкоцитов. Предполагают, что благодаря этим совместным эффектам (а возможно, и другим) вазоокклюзионные кризы удается купировать.

- Рекомендуем ознакомиться со следующей статьей "Механизмы развития (патогенез) бета-талассемии"
Оглавление темы "Патогенез анемий":- Механизмы развития (патогенез) серповидно-клеточной анемии
- Механизмы развития (патогенез) бета-талассемии
- Механизмы развития (патогенез) альфа-талассемии
- Механизмы развития (патогенез) пароксизмальной ночной гемоглобинурии
- Механизмы развития (патогенез) иммуногемолитической анемии
- Механизмы развития (патогенез) мегалобластной анемии
- Механизмы развития (патогенез) пернициозной анемии
- Механизмы развития (патогенез) анемии при дефиците фолиевой кислоты
- Механизмы развития (патогенез) железодефицитной анемии
- Механизмы развития (патогенез) анемии из-за хронического заболевания