
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Схема поражения двигательной единицы, миотонии и мышечной дистрофии
Двигательная единица состоит из мотонейрона (а-мотонейрон в спинном мозге или черепно-мозговых нервах), его аксона и всех мышечных волокон, иннервируемых его коллатералями. Нарушение функции двигательной единицы может быть обусловлено поражением мотонейрона, прерыванием или замедлением аксональной проводимости либо патологией мышцы.
Вирус полиомиелита может инфицировать а-мотонейроны и необратимо разрушать их. Также эти клетки разрушаются при спинальной мышечной атрофии (разнородная группа дегенеративных заболеваний). Генетические дефекты СОД, которая в норме защищает нейроны от окислительного стресса, вызывают боковой амиотрофический склероз. Недостаточная активность СОД приводит к гибели спинальных а-мотонейронов и супраспинальных мотонейронов.
Встречаются мутации, которые ведут к дефектам динактина (аксональный транспорт), митохондриальной цитохром с-оксидазы и гена ALSIN (регуляция эндосомного транспорта). При передаваемом по наследству Х-сцепленном синдроме Кеннеди гибель а-мотонейронов происходит из-за дефекта рецепторов андрогенов.
Повреждение или гибель аксонов помимо прочего могут вызывать аутоиммунные заболевания, недостаток витамина В1 или В12, сахарный диабет, интоксикацию (например, свинцом, алкоголем) или генетические дефекты (например, синдром Шарко—Мари—Тута).
Скелетные мышцы могут повреждаться при аутоиммунных заболеваниях (например, дерматомиозите), а также вследствие генетических дефектов, например, при миотонии или дистрофии.
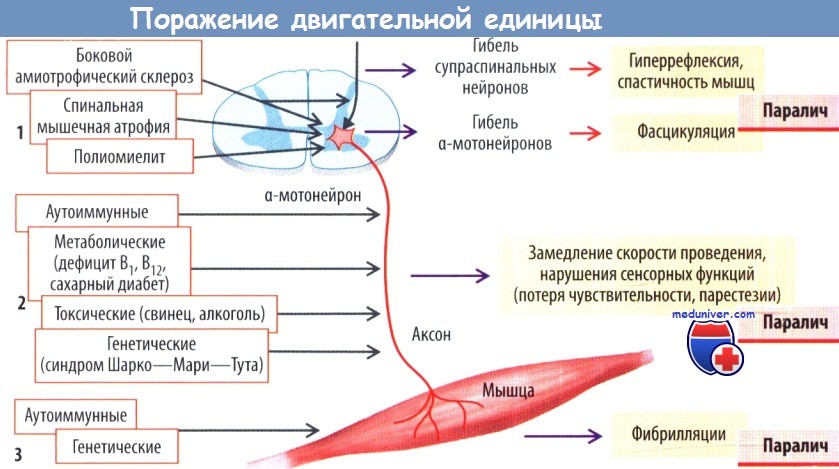
Повреждение двигательной единицы ведет к параличу мышц независимо от того, локализуется ли очаг поражения в а-мотонейроне, аксоне либо в самой мышце. При гибели а-мотонейрона, как правило, наблюдаются фасцикуляции. Они происходят в результате одновременной стимуляции и сокращения мышечных волокон двигательной единицы. При боковом амиотрофическом склерозе разрушение супраспинальных нейронов может приводить к гиперрефлексии и спастичности мышц, пока некоторые а-мотонейроны остаются интактными.
Повреждение периферического нерва, при котором уменьшается толщина миелинового слоя, обусловливает снижение скорости проведения импульсов.
Чувствительная часть нерва, как правило, тоже поражается. Это сопровождается нарушением чувствительности, а также возникновением спонтанных ПД в поврежденном нерве, что вызывает соответствующие ощущения (парестезии). Если происходит гибель самой мышцы, обычно наблюдаются фибрилляции, т. е. некоординированные сокращения отдельных мышечных волокон.
Генетические дефекты ионных каналов служат причиной развития целой группы функциональных заболеваний мышц. Обычно клеточная мембрана мышечной клетки деполяризуется в ответ на возбуждение потенциалзависимого Na+-канала, что вызывает открытие потенциалзависимого Са2+-канала и Са2+-канала саркоплазматического ретикулума.
В результате этих процессов внутриклеточная концентрация Са2+ увеличивается, вызывая мышечное сокращение. Реполяризация достигается путем инактивации Na+-каналов, притока Cl- и выхода К+ из клетки. Это запускает инактивацию Са2+-каналов, поэтому внутриклеточная концентрация Са2+ вновь снижается и мышца расслабляется.
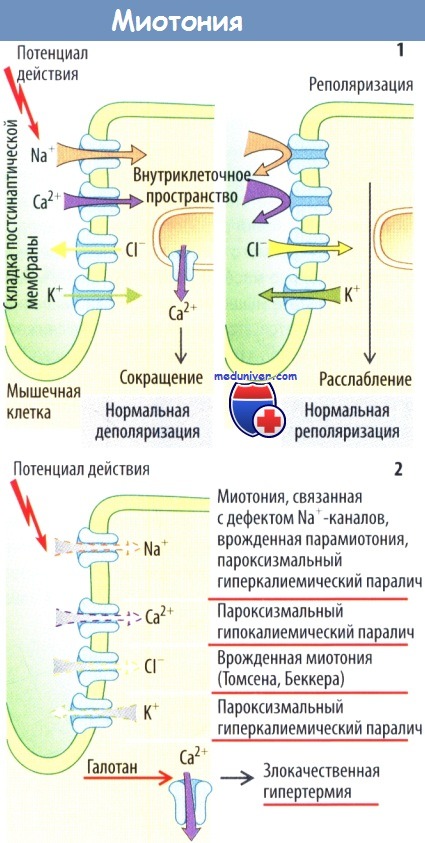
Запаздывание инактивации Na+-каналов из-за мутации гена, кодирующего белок ионного канала, может приводить к задержке расслабления мышцы, повышенной возбудимости и судорогам (миотония, связанная с дефектом Na'-каналов, и врожденная парамиотония). Холод способствует замедлению инактивации Na+-каналов, что ведет к судорогам, особенно при парамиотонии.
Наличие дополнительного дефекта Na+-канала или дефектного К+-канала может вызывать паралич вследствие высокой внеклеточной концентрации К+ (гиперкалиемический пароксизмальный паралич). Генетический дефект потенциалзависимого Са2+-канала также приводит к гипокалиемическому пароксизмальному параличу. Дефекты Cl--каналов вызывают миотонию. Эти каналы состоят из нескольких субъединиц.
Если включение измененной субъединицы нарушает функцию всего комплекса, такая мутация характеризуется доминантным наследованием (врожденная миотония, болезнь Томсена). Если сама субъединица нефунк циональна, но не нарушает функцию интактных субъединиц, мутация характеризуется рецессивным наследованием (миотония Беккера). При определенных дефектах Са2+-каналов саркоплазматического ретикулума (рианодиновый рецептор) галогенизированные анестетики могут вызывать потенциалнезависимую активацию этих каналов с увеличением количества внутриклеточного Са2+. Это приводит к существенному повышению энергетического обмена и развитию гипертермии (злокачественная гипертермия).
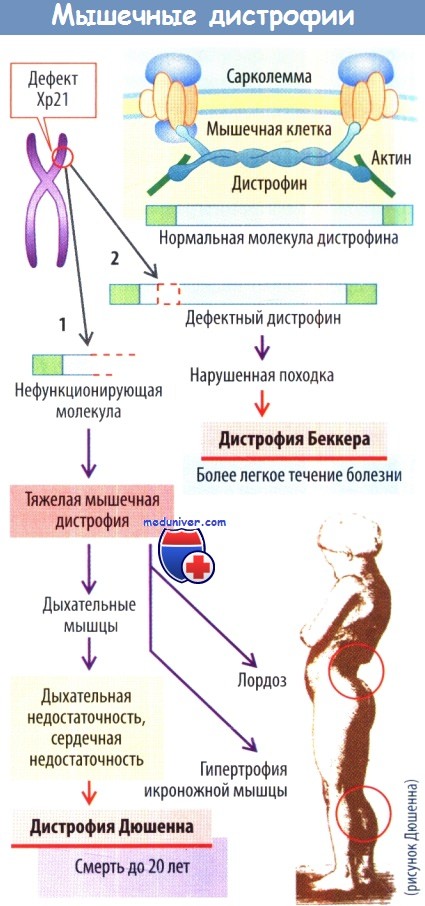
При дегенеративной мышечной дистрофии Дюшенна или Беккера поврежден дистрофии, один из элементов цитоскелета. Ген, кодирующий этот белок, находится на хромосоме X. Это заболевание встречается фактически только у мужчин, т.к. у женщин с одним дефектным геном образуется достаточное количество дистрофина, кодируемого нормальным геном. При дистрофии Дюшенна формируются лишь короткие, совершенно нефункциональные фрагменты дистрофина. Это заболевание заканчивается смертью в течение первых 20 лет жизни.
Для этой формы дистрофии типичны выраженный поясничный лордоз позвоночника и гипертрофированные, но, несмотря на это, слабые икроножные мышцы. Вовлечение в патологический процесс кардиомиоцитов приводит к кардиомиопатии. При дистрофии Беккера дистрофии также дефектный, но его функция страдает в меньшей степени, поэтому течение заболевания не столь неблагоприятное, как при дистрофии Дюшенна.
Причиной мышечных дистрофий могут служить дефекты следующих мышечных белков: миотилин, ламин.кавеолин, калпаин, дисферлин, саркогликан, телетонин и титин. Кроме того, миопатии могут быть следствием метаболических дефектов (например, гликогенозов), эндокринных нарушений (например, гипертиреоза) или аутоиммунных заболеваний (например, полимиозита, дерматомиозита).
- Рекомендуем ознакомиться со следующей статьей "Схема электромиографии и образования креатинина"
Оглавление темы "Патофизиология в схемах":- Схема последствий избытка инсулина и развития гипогликемии
- Схема функций гистамина и брадикинина
- Схема функций серотонина
- Схема функций эйкозаноидов
- Схема вариантов повреждений нервной системы
- Схема болезней клеток нервной системы - нейронов
- Схема развития демиелинизации нервов при рассеянном склерозе
- Схема нарушения нервно-мышечной передачи при миастении и синдроме Ламберта-Итона
- Схема поражения двигательной единицы, миотонии и мышечной дистрофии
- Схема электромиографии и образования креатинина