
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Ген p53 и его значение в развитии опухоли
Ген р53 — страж генома. Ген р53 расположен на хромосоме 17р13.1 и является наиболее частой мишенью генетических повреждений в опухолях человека. Официальное название гена ТР53, а его белкового продукта — р53; с целью упрощения изложения материала далее оба будем называть р53. Около 50% опухолей человека ассоциированы с мутацией в этом гене.
Гомозиготная утрата р53 происходит практически во всех видах злокачественных опухолей, включая карциномы легкого, толстой кишки и молочной железы — три лидирующие онкологические причины летального исхода. В большинстве случаев инактивирующая мутация затрагивает оба аллеля р53 и развивается в соматических клетках (не в половых клетках). Реже некоторые лица наследуют мутантный аллель р53.
Как в случае с геном RB, наследование одного мутантного аллеля предрасполагает к развитию злокачественных опухолей, поскольку достаточно только одного дополнительного удара для инактивации второго, нормального аллеля. Эту болезнь называют синдромом Ли-Фраумени. При его наличии у пациента в 25 раз повышается риск развития злокачественной опухоли до 50 лет по сравнению с общей популяцией.
В отличие от пациентов, которые наследуют мутантный аллель RB, спектр опухолей у пациентов с синдромом Ли-Фраумени включает самые распространенные новообразования — саркомы, рак молочной железы, лейкемии, опухоли головного мозга и карциномы коры надпочечников. По сравнению со спорадическими опухолями злокачественные новообразования у пациентов с синдромом Ли-Фраумени манифестируют в более молодом возрасте, у таких пациентов также могут развиваться первично-множественные опухоли.
Частое выявление мутаций р53 в разнообразных опухолях позволяет предположить, что белок р53 выполняет функцию стража генома и защищает организм от развития злокачественных опухолей, т.е. работает «молекулярным полицейским», предотвращая размножение клеток с поврежденным геномом. р53 — фактор транскрипции, являющийся центральным участником разветвленной сети сигналов, регистрирующих стрессовые состояния клетки, например повреждение ДНК, укорочение теломераз и гипоксию.
Многие стороны активности белка р53 связаны с его деятельностью как фактора транскрипции. Было установлено: несколько сотен генов, отвечающих за множество разнообразных процессов, регулируются р53. Но какой из них является ключевым для р53, остается неясным. Около 80% точечных мутаций, обнаруженных в злокачественных опухолях человека, располагаются в домене р53, связывающем ДНК.
В то же время эффект разных точечных мутаций чрезвычайно разнообразен: в некоторых случаях это полная утрата транскрипционной способности, в других — связывание и активация подмножества генов. Функционирование р53 может быть инактивировано не только приобретенными соматическими и наследственными мутациями, но и с помощью других механизмов. Как и с белком RB, нормальный р53 также может быть инактивирован трансформирующими белками определенных ДНК-вирусов (например, белком Е6 HPV), которые могут связываться с р53 и деградировать его. Предполагают, что в подавляющем большинстве опухолей без идентификации мутаций р53 все равно присутствует блокада сигнального пути р53 другими генами, регулирующими функционирование р53. Так, MDM2 и MDMX стимулируют деградацию р53; в злокачественных опухолях без мутаций р53 часто отмечается повышение экспрессии этих генов. Действительно, MDM2 амплифицирован в 33% сарком, что ассоциировано с утратой функциональной активности р53 в данных опухолях.
р53 препятствует злокачественной трансформации клеток тремя взаимосвязанными механизмами: путем активации временной остановки клеточного цикла и вывода клетки из него (это называется состоянием покоя), индукции постоянной остановки цикла и вывода клетки из него (это называется состоянием старения) или индукции запрограммированной смерти клетки (апоптоза).
В неповрежденных клетках р53 является короткоживущей молекулой (20 мин) из-за ее ассоциации с MDM2, белком, который предназначен для разрушения р53. Когда клетка находится в состоянии стресса и есть повреждения ее ДНК, р53 подвергается посттранскрипционным модификациям, которые освобождают его от MDM2, что увеличивает период жизни р53 и активирует как фактор транскрипции. Найдено множество генов, транскрипция которых вызвана р53. Эти гены можно сгруппировать в две большие категории: те, которые вызывают остановку клеточного цикла, и те, которые приводят к апоптозу. Если повреждения ДНК могут быть устранены во время остановки клеточного цикла, клетка возвращается в нормальное состояние; если «ремонт» ДНК невозможен, р53 вызывает старение или апоптоз клетки. Список регуляторных молекул был расширен. Было установлено, что подавление группы пролиферативных и антиапоптозных генов является ключевым эффектом, который оказывает влияние на ответ р53.
При этом остается непонятным, почему р53 подвергается репрессии, в то время как в большинстве случаев он активирует транскрипцию. В связи с этим обратимся к получившим не так давно известность микроРНК. Установлено, что р53 активирует транскрипцию семейства mir34 микроРНК (mir34a, mir34b, mir34c). МикроРНК (см. главу 5) присоединяется к родственным последовательностям в 3'-нетранслируемых участках мРНК, предотвращая их трансляцию. Интересно, что блокирование mir34 значительно нарушает ответ р53, в то время как индуцированной извне экспрессии mir34, не сопровождающейся активацией р53, достаточно для остановки роста и апоптоза. Следовательно, mir34 микроРНК способна повторять многие функции р53 и необходима для их выполнения. Мишенями для mir34 являются пролиферативные гены, например циклины, и антиапоптозные гены — Bcl-2 и другие. Регулирование р53 посредством mir34 отчасти объясняет, как р53 репрессирует экспрессию генов, а также то, что микроРНК играет критическую роль в ответе р53.
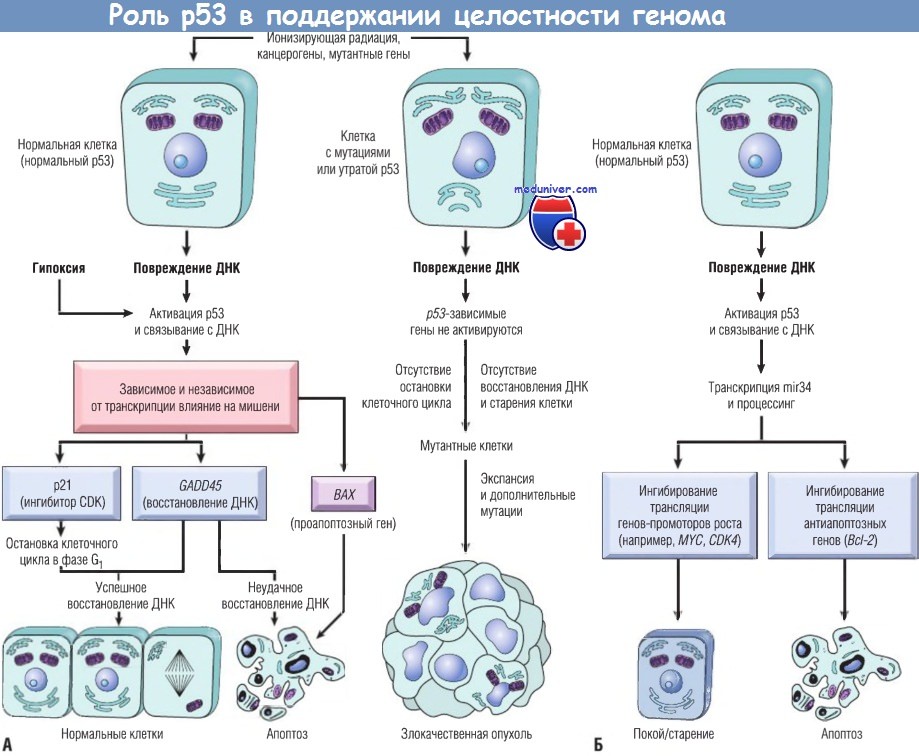
Активация нормального р53 агентами, повреждающими ДНК, а также гипоксией приводит к остановке клеточного цикла в фазе G1 и индукции восстановления ДНК путем повышения транскрипции р21 (CDKN1A) и гена GADD45.
Успешное восстановление ДНК позволяет клеткам вновь входить в митотический цикл; если восстановление ДНК неудачно, р53 инициирует апоптоз или старение клетки.
В клетках с утратой или мутациями р53 остановки клеточного цикла или восстановления ДНК при ее повреждении не происходит, генетически поврежденные клетки пролиферируют и подвергаются в конечном счете злокачественной трансформации.
(Б) р53-индуцированная репрессия генов путем активации микроРНК. р53 активирует транскрипцию mir34 семейства микроРНК.
mir34 подавляет транскрипцию как пролиферативных генов, например циклинов, так и антиапоптозных генов, например Всl-2.
Подавление этих генов может усиливать как вхождение клеток в состояние покоя или старения, так и апоптоз.
CDK — циклин-зависимые киназы; ДНК — дезоксирибонуклеиновая кислота.
Как р53 удается определить повреждение ДНК и адекватность ее восстановления, остается непонятным. Ключевыми инициаторами повреждения ДНК являются две взаимосвязанные протеинкиназы: мутантная атаксии-телеангиэктазии (ATM) и атаксии-телеангиэктазии и Rad3 (ATR). В название гена ATM отражена первоначальная идентификация его мутации в половых клетках у пациентов с синдромом атаксиителеангиэктазии. Организм пациентов с этой болезнью не способен устранить определенные виды повреждений ДНК, больные часто страдают от злокачественных опухолей. Типы повреждений, определяемых ATM и ATR, различны, но регуляторные пути, которые они активируют, сходны.
После активации ATM и ATR фосфорилируют многие молекулы, включая р53 и белки восстановления ДНК. Фосфорилирование этих двух молекул приводит к паузе в клеточном цикле и восстановлении ДНК соответственно.
р53-опосредованную остановку клеточного цикла можно считать первичным ответом на повреждение ДНК. Остановка происходит на позднем этапе фазы G1 и вызвана главным образом р53-зависимой транскрипцией ингибитора CDK — CDKN1A (р21). Ген р21 блокирует комплексы циклин-CDK и препятствует фосфорилированию RB, что необходимо для входа клетки в фазу G1. Такая остановка клеточного цикла является долгожданным моментом, т.к. дает клетке «перевести дыхание» для восстановления ДНК.
р53 также вызывает экспрессию определенных белков, участвующих в восстановлении ДНК, например GADD45, что помогает процессу восстановления ДНК. Помимо этого р53 может стимулировать регуляторные пути восстановления ДНК, независимые от механизмов транскрипции. Если повреждения ДНК успешно устранены, то р53 стимулирует транскрипцию MDM2 и соединяется с ним, что приводит к разрушению р53 и снятию блокады клеточного цикла. Если повреждения устранить невозможно, то клетка может подвергнуться р53-индуцированному старению или р53-опосредованному апоптозу.
р53-индуцированное старение — постоянная остановка клеточного цикла, характеризующаяся определенными изменениями в морфологии и экспрессии генов, отличающимися от таковых в состоянии покоя или временной остановки клеточного цикла. Для старения клеток необходима активация р53 и/или RB и экспрессия их посредников, например CDKI. Механизмы старения неясны, но, вероятно, сопряжены с глобальными изменениями хроматина, которые существенно и надолго нарушают экспрессию генов.
Локусы гетерохроматина, ассоциированные со старением клеток, содержат пролиферативные гены, регулируемые E2F. Подобные изменения вызывают выраженное и необратимое нарушение экспрессии мишеней E2F. Старение, как и другие р53-опосредованные эффекты, может быть обусловлено разнообразными стрессорными факторами, такими как беспрепятственная экспрессия онкогенов, гипоксия и укорочение теломер.
р53-индуцированный апоптоз клеток с необратимым повреждением ДНК — наивысшая точка антибластомной резистентности клетки. р53 запускает транскрипцию с помощью нескольких проапоптозных генов, например ВАХ и PUMA (общепринятое название ВВС3). Точно не установлено, каким образом клетка принимает решение, восстанавливать ли ей поврежденную ДНК или погибнуть путем апоптоза. Как оказалось, способность р53 стимулировать и поддерживать гены восстановления ДНК намного выше, чем сродство к проапоптозным генам. Следовательно, сигнальный путь восстановления ДНК запускается первым, а р53 продолжает накапливаться в клетке. В результате к моменту, когда становится очевидной невозможность устранить повреждение ДНК, в клетке уже присутствует достаточное количество р53, позволяющее запустить апоптоз, и клетка погибает. Хотя данная схема в целом верна, но, вероятно, существуют специфические ответы клеток, когда одни гораздо раньше подвергаются апоптозу, а другие — старению.
Подобные различия в ответных реакциях могут быть связаны с функционированием других членов семейства р53, которые экспрессируются в различных клетках.
Следует подчеркнуть, что ген р53 называют «стражем генома», т.к. он обеспечивает взаимосвязь между повреждением и его устранением ДНК, выход клетки из митотического цикла и ее апоптоз. Восстановление ДНК осуществляется путем остановки клеточного цикла в фазе G1 и активации генов восстановления ДНК. Клетка с повреждением ДНК, которое невозможно устранить, подвергается апоптозу. При утрате функциональной активности р53 восстановления поврежденной ДНК не происходит, мутации в делящихся клетках закрепляются и клетка «переходит на улицу с односторонним движением», приводящую ее к злокачественной трансформации.
Способность р53 индуцировать апоптоз в ответ на повреждение ДНК имеет важное значение для лечения опухолей. Облучение и химиотерапия, два основных метода лечения злокачественных опухолей, основаны на индукции повреждения ДНК и последующего апоптоза клеток. Опухоли с нормальным р53 лучше отвечают на эту терапию, чем содержащие мутантные аллели гена, как, например, в случаях тератокарциномы яичка и острой лимфобластной лейкемии у детей. Напротив, опухоли при раке легкого и колоректальном раке, имеющие часто мутантный р53, являются относительно резистентными к химиотерапии и облучению. Различные методы терапии направлены на поддержание и усиление активности нормального р53, а также на поиск способов селективного киллинга клеток с нарушением функции р53.
Обнаружение других членов семейства р53, например р63 и р73, свидетельствует, что р53 имеет помощников. Действительно, существует сложная сеть с перекрестным обменом информацией между р53, р63 и р73. Эту сеть начали изучать и должны «распутать». р53 экспрессируется в большинстве опухолей, тогда как р6З и р73 обладают выраженной тканеспеци-фичностью. Например, р6З необходим для плоскоклеточной дифференцировки, а р73 обладает выраженным проапоптозным эффектом в ответ на повреждение ДНК в результате химиотерапии. Более того, р63, р73 и, вероятно, р53 существуют в нескольких изоформах, часть которых работают как активаторы транскрипции, другие, напротив, как доминантные ингибиторы. Проиллюстрировать их совместный эффект можно на примере базально-клеточного рака молочной железы, имеющего плохой прогноз. В этих опухолях одновременно присутствуют мутантный р53 и экспрессия доминантно-негативной изоформы р63, которая противодействует апоптозной активности р73. Нарушения в системе р53-р63-р73 обусловливают химиорезистентность и плохой прогноз.
- Рекомендуем ознакомиться со следующей статьей "Ген APC и его значение в развитии опухоли"
Оглавление темы "Патофизиология онкологических заболеваний":- Онкоген RAS и его характеристика
- Изменения нерецепторных тирозинкиназ при развитии рака
- Протоонкоген MYC в развитии опухолей
- Циклины и циклин-зависимые киназы в развитии опухолей
- Гены-супрессоры опухоли и их характеристика
- Ген RB и его значение в развитии опухоли
- Ген p53 и его значение в развитии опухоли
- Ген APC и его значение в развитии опухоли
- Ген INK4a/AKF (локус CDKN2A) и его значение в развитии опухоли
- Сигнальный путь TGF-b и его значение в развитии опухоли