
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Тирозинемия у новорожденных - причины, диагностика, лечение
Тирозинемия - редкое заболевание, связанное с нарушениями аминокислотного обмена. Разновидности тирозинемии последовательно описаны следующими исследователями: M.D. Baber (1956) — тирозинемия тип I; Н. Richner (1938) и Е. Hanhart (1947) — тирозинемия тип II; О. Giardini и соавт. (1983), F. Endo и соавт. (1983) — тирозинемия тип III.
Распространенность тирозинемии тип I составляет порядка 1 случая на 50 000-100 000 новорожденных; тирозинемия II типа — нечастая патология (частота встречаемости не изучена), а III тип болезни исключительно редок.
Тирозинемия — наследственное заболевание обмена веществ, связанное с нарушениями обмена тирозина, приводящее, в зависимости от типа болезни, к преимущественному поражению ЦНС, печени, почек, зрения, кожных покровов и умственной отсталости.
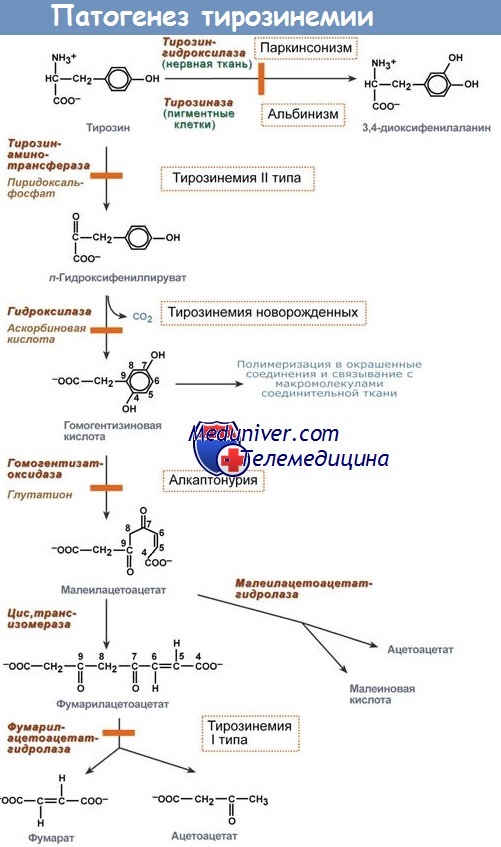
Нарушения метаболизма при различных формах тирозинемии детерминированы нарушениями работы ферментов: фумарилацетатацетазы, тирозинтрансаминазы, 4-гидроксифенил пируватдиоксидазы (ГФПД).
Классификация тирозинемии
В настоящее время известны не менее шести вариантов тирозинемии: тирозинемия тип I (наследственная или гепаторенальная тирозинемия — следствие дефекта фумарилацетоацетатгидролазы) — острая или хроническая; тирозинемия тип II (синдром Ричнера-Ханхарта) — вследствие дефекта растворимой формы тирозинтрансаминазы; тирозинемия тип III — результат дефекта 4-гидроксифенилпируватдегидрогеназы (ГФПД); тирозиноз — казуистически редкое асимптоматическое состояние, сопровождаемое гипертирозинемией (энзиматический дефект неизвестен); транзиторная тирозинемия недоношенных новорожденных.
Последняя возникает вследствие инактивации ГФПД ее субстратом; по-видимому, не является врожденной ошибкой метаболизма, а лишь отражает реакцию организма на интенсивное потребление белка.
Существует также хоукинсинурия (hawkinsinuria) — состояние, характеризующееся экскрецией цистеина или глутатион-конъюгированного продукта интермедиарного обмена в процессе конверсии 4-гидроксифенилпировиноградной кислоты (ГФПК) в гомогенизиновую кислоту.
Генетические аспекты тирозинемии
Как и при большинстве заболеваний из группы нарушений аминокислотного обмена, для тирозинемии (типов I, II и III) характерен аутосомно-рецессивный тип наследования, т.е. 25% детей двух клинически здоровых носителей (гетерозигот) оказываются пораженными.
Ген тирозинемии тип I (OMIM #276700) локализуется на хромосоме 15 (регион 15q23-25), тирозинемиии тип II — на длинном плече хромосомы 16 (локус 16q22.1-q22.3).
Молекулярно-генетические особенности различных типов тирозинемии (I, II и III) в настоящее время изучаются.
Клинические проявления и диагностика тирозинемии
Тирозинемия тип I определяется недостаточностью фумарилацетоацетазы. При этом происходит токсическое накопление фумарилацетоацетата и малеилацетоацетата в различных тканях органах организма.
При острой (неонатальной) форме болезни присутствуют признаки нейротоксикоза, мышечной гипотонии, отмечается повышение температуры тела (до фебрильных значений), имеют место затяжное течение желтухи, гепатоспленомегалия, сопровождаемая рвотой, диареей и обезвоживанием. У детей отмечается выраженное отставание в физическом развитии (гипотрофия), на фоне чего возникновение инфекционных заболеваний приводит к провокации острых печеночных кризов, желудочно-кишечных кровотечений и развитию асцита. Прогноз острой формы болезни чрезвычайно неблагоприятен (цирроз печени, печеночная недостаточность, карцинома печени).
Существует также хроническая форма тирозинемии тип I (с более поздней манифестацией симптомов и более легким течением, но с острыми приступами периферической полинейропатии).
В диагностике тирозинемии специфичны следующие основные изменения биохимических показателей: в моче — резкое увеличение содержания тирозина на фоне общей гипераминоацидурии, повышение концентраций сукцинилацетона, пара-гидроксифениллактата, пара-гидроксифенилпирувата, пара-гидроксифенилацетата; снижение активности фумарилацетоацетазы в лимфоцитах и биоптате печени; в крови — резкое увеличение концентрации тирозина (в 20-50 раз) на фоне общей гипераминоацидемии (повышаются также уровни фенилаланна и метионина).
Среди неспецифических изменений биохимических показателей крови отмечаются следующие: гипербилирубинемия, повышение активности ферментов печени (АЛТ, ACT), гиперхолес-теринемия.
МРТ-исследование головного мозга при тирозинемии тип I может выявить двусторонние изменения высокого сигнала в области бледного ядра на Т2-взвешенных изображениях, что сопровождается явлениями астроцитоза, задержкой миелинизации и status spongiosus (расщепление и вакуолизация миелина).
Жидкостная хроматография/масс-спектрометрия при проведении скрининга новорожденных для выявления тирозинемии тип I нередко дает ложноотрицательные результаты, в связи с чем G. 1а Магса и соавт. (2008) предлагают в качестве маркера болезни использовать сукцинилацетон.
Тирозинемия тип II (синдром Ричнера-Ханхарта) — заболевание, при котором возраст пациента ко времени появления клинической симптоматики чрезвычайно вариабелен (от неонатального до подросткового периода), хотя чаще первые признаки болезни возникают между 1 и 4 годами. Помимо умственной отсталости и задержки физического развития, отличительными чертами этого варианта болезни являются гиперкератоз и эрозии (буллезные) на ладонях и подошвах стоп, а также (до 75% случаев) различные поражения глаз: снижение остроты зрения, конъюнктивит, изъязвление роговицы и др.
Поражение глаз сопряжено с тем, что в результате метаболического блока тирозинаминотрансферазы в биологических тканях и жидкостях аккумулируется тирозин, приводя к отложению кристаллов в роговичной оболочке.
Неврологическая симптоматика наличествует у подавляющего большинства детей (вялость, сонливость, нарушения мелкой моторики, дефекты речи и т.д.). Подчеркивается необходимость максимально раннего установления у детей диагноза тирозинемии тип II во избежание риска формирования умственной отсталости.
Тирозин — единственная аминокислота, содержание которой повышается в моче у детей с этим типом болезни (уровни содержания других аминокислот остаются нормальными); нарастает также содержание в моче 4-гидроксифенилпировиноградной, 4-гидроксифенилмолочной, 4-гидроксифенилуксусной и фенилуксусной кислот, а также N-ацетилтирозина. Разработан ряд методов молекулярно-генетической диагностики тирозинемии тип II.
Тирозинемия тип III — вариант болезни, проявляющийся на 1-м году жизни (обычно по истечении неонатального периода) с манифестацией в виде неврологической симптоматики (судорожный синдром — по типу инфантильных спазмов; атаксия, гемипарезы, вялость, мышечная гипотония, задержка психомоторного развития), а также признаков поражения других органов и систем (гепатомегалия, расстройства дыхания, в более старшем возрасте может выявляться тиреоидит).
При тирозинемии тип III в крови и моче регистрируется высокое содержание тирозина и его метаболитов; отмечается выраженная гипераммониемия с умеренной гиперфенилаланинемией. В биоптатах печени определяется снижение активности 4-гидроксифенилпируватдиоксигеназы.
Транзиторная тирозинемия недоношенных детей — патология, которая, несмотря на название, встречается не только у недоношенных новорожденных. Клинически проявляется затяжным течением желтухи, анорексией и вялостью. Впоследствии возможны задержка психомоторного развития и интеллектуальный дефицит.
Хоукинсинурия — доброкачественное транзиторное состояние, зарегистрированное во всем мире лишь в нескольких семьях (впервые в 1977 г.). Помимо собственно гипертирозинемии, хоукинсинурия характеризуется метаболическим ацидозом и неспособностью ребенка к прибавке массы тела. Хоукинсинурия прогностически благоприятна.
Профилактика тирозинемии
Максимально раннее выявление болезни (включая дородовую диагностику тирозинемии тип I — используется определение содержания сукцинилацетона в амниотической жидкости и анализ активности фумарилацетоацетазы в культивированных клетках или ворсинках хориона; для выявления тирозинемии типов I и II, а также части случаев тирозинемии тип III применяется ДНК-анализ).
Лечение тирозинемии
За рубежом имеется опыт применения при лечении тирозинемии тип I препарата NTBC (2-нитро-4-трифлюорометилбензоил-1,3-циклогексанедион), препятствующего выработке фумарилацетоацетата. В США NTBC введен в рутинную практику с 2002 г. Под названием нитизинон {nitisinone) препарат является первым лекарственным средством, одобренным в Европе для лечения наследственной тирозинемии тип I. Подчеркивается, что NTBC эффективен при выявлении заболевания у детей в течение первого полугодия жизни. М. Ashom и соавт. (2008) сообщают об эффективности применения NTBC в Финляндии, a A. Masurel-Paulet и соавт. (2008) — во Франции. С 2009 года нитизинон (орфадин*) зарегистрирован и доступен в РФ.
Другие методы терапии включают использование гемодиализа и трансплантацию печени. Остальные лечебные методики являются симптоматическими.
Основой лечения болезни считается диетотерапия (как на фоне применения NTBC при типе I, так и без такового).
- Вернуться в оглавление раздела "неврология"
Оглавление темы "Неврология новорожденных детей":- Синдром ригидного младенца (stiff baby syndrome) - причины, диагностика, лечение
- Синдром Шварца-Джампеля - причины, диагностика, лечение
- Врожденная (семейная) дисавтономия (синдром Райли-Дэя) - причины, диагностика, лечение
- Гликогеноз тип IIa (болезнь Помпе) - причины, диагностика, лечение
- Инфантильные периферические нейропатии - причины, диагностика, лечение
- Фенилкетонурия (ФКУ) у новорожденных - причины, диагностика, лечение
- Болезнь мочи с запахом кленового сиропа (БМЗКС, лейциноз) у новорожденных - причины, диагностика, лечение
- Галактоземия у новорожденных - причины, диагностика, лечение
- Гистидинемия у новорожденных - причины, диагностика, лечение
- Тирозинемия у новорожденных - причины, диагностика, лечение