
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Фенилкетонурия (ФКУ) у новорожденных - причины, диагностика, лечение
Врожденные ошибки метаболизма (англ. inborn errors/defects of metabolism) — условный термин, объединяющий гетерогенную группу генетически-детерминированных заболеваний (аминоацидурии, органические ацидурии/ацидемии, дефекты обмена аминокислот, углеводов и т.д.) с первичным или вторичным поражением нервной системы.
Фенилкетонурия (ФКУ) — наследственное заболевание, характеризующееся нарушениями обмена фенилаланина. Классический вариант ФКУ описал Asbjorn Foiling (1934).
Фенилкетонурия (ФКУ) встречается с частотой 1 случай на 8000-15 000 новорожденных (в странах Европы — 1 на 10000 новорожденных).
Выделяют четыре основные формы ФКУ, хотя существует более 400 различных мутаций и несколько метаболических фенотипов ФКУ
Фенилкетонурия — заболевание, относящееся к группе наследственных аминоацидопатий, связанное с нарушением метаболизма фенилаланина, в результате мутационной блокады работы ферментов приводящее к стойкой хронической интоксикации и поражению ЦНС, выраженному снижению интеллекта и неврологическому дефициту различной степени выраженности.
Этиология и патогенез фенилкетонурии
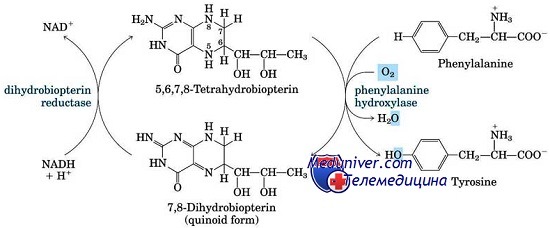
Как известно, фенилаланин (ФА) — это эссенциальная аминокислота, которая в норме при поступлении с диетой не утилизируется для белкового синтеза, а гидроксилируется до тирозина, фенилаланин-4-гидроксилаза (ФАГ) — фермент, обеспечивающий трансформацию фенилаланина в тирозин.
Основное значение в патогенезе классической фенилкетонурии придается неспособности фенилаланин-гидроксилазы перерабатывать фенилаланин до тирозина, в результате чего в организме накапливаются фенилаланин и продукты его аномального обмена (фенилпировиноградная, фенилук-сусная, фенилмолочная кислоты).
В числе других патогенетических факторов в настоящее время рассматриваются следующие: нарушения аминокислотного транспорта через гематоэнцефалический барьер (ГЭБ); нарушения церебрального пула аминокислот с последующим нарушением синтеза протеолипидных белков; нарушения миелинизации; низкие уровни нейротрансмиттеров (серотонин и др.).
Фенилкетонурия (ФКУ) I (классическая или тяжелая) — аутосомно-рецессивное заболевание, вызванное мутацией гена фенилаланингидроксилазы, локализованного в длинном плече хромосомы 12 (выявлено 12 различных гаплотипов, из которых около 90% ФКУ ассоциировано с 4 гаплотипами). Наиболее частые мутации в гене фенилаланин гидроксилазы: R408W, R261Q, IVS10 nt 546, Y414C.
В основе болезни лежит дефицит фенилаланин-4-гидроксилазы, обеспечивающей конверсию фенилаланина в тирозин, что приводит к накоплению в тканях и физиологических жидкостях больного фенилаланина и его метаболитов.
Особую группу составляют атипичные варианты ФКУ При них клиническая картина напоминает классическую форму ФКУ, но по показателям развития, несмотря на проведение диетотерапии, не отмечается положительной динамики. Атипичные варианты ФКУ связаны с дефицитом дегидроптеринредуктазы, тетрагидроптерина, 6-пирувоилтетрагидроптеринсинтазы, гуанозин-5-трифосфатциклогидролазы и т.д.
Фенилкетонурия (ФКУ) II (атипичная) — аутосомно-рецессивное заболевание, при котором генный дефект локализуется в коротком плече хромосомы 4 (участок 4р15.3).
ФКУ II характеризуется недостаточностью дегидроптеринредуктазы, приводящей к нарушению восстановления активной формы тетрагидробиоптерина (кофактора в гидроксилировании фенилаланина, тирозина и триптофана) в сочетании со снижением в сыворотке крови и СМЖ фолатов. Результатом являются метаболические блоки в механизмах превращения фенилаланина в тирозин, а также предшественников нейромедиаторов катехоламинового и серотонинового рядов (L-дофа, 5-окситриптофан), о чем свидетельствует резкое снижение концентрации содержания в тканях и физиологических жидкостях пациентов их конечных продуктов (гомованилиновой и 5-оксииндолуксусной кислот). Болезнь описана I. Smith (1974), затем S. Kaufman и соавт. (1975) обнаружили дефицит дегидроптеринредуктазы при ФКУ II.
Фенилкетонурия (ФКУ) III (атипичная) — аутосомно-рецессивное заболевание, связанное с недостаточностью 6-пирувоилтетрагидроптеринсинтазы, участвующей в процессе синтеза тетрагидробиоптерина из дигидронеоптерин трифосфата (описали S. Kaufman и соавт., 1978). Вследствие дефицита тетрагидробиоптерина у пациентов отмечаются расстройства, сходные с нарушениями при ФКУ П.
Примаптеринурия — еще один вариант атипичной ФКУ у детей с легкой ГФА, когда в моче в больших количествах присутствуют примаптерин и некоторые его производные при наличии нормальной концентрации в спинномозговой жидкости нейромедиаторных метаболитов (гомованилиновой и 5-оксииндолуксусной кислот). Энзиматический дефект при указанной форме атипичной ФКУ пока не выявлен.
Материнская фенилкетонурия (ФКУ) — заболевание, сопровождающееся снижением интеллекта (до умственной отсталости) среди потомства женщин, страдающих ФКУ и не получающих специализированную диету в совершеннолетнем возрасте.
Патогенез материнской фенилкетонурии детально не изучен, но предполагается роль хронической интоксикации плода фенилаланином и продуктами его аномального метаболизма. R. Koch и соавт. (2008) при аутопсии головного мозга младенца, у матери которого отмечалась ФКУ (без адекватного контроля за уровнем фенилаланина в крови), обнаружили ряд патологических изменений: низкую массу мозга, вентрикуломегалию, гипоплазию белого вещества и задержку миелинизации в путях поздней миелинизации (без признаков астроцитоза); хронических изменений в сером веществе головного мозга не было обнаружено.
Предполагается, что именно нарушения в развитии белого вещества мозга ответственны за формирование неврологического дефицита при материнской фенилкетонурии.
Варианты классификации фенилкетонурии (ФКУ)
В классификации ФКУ по Е. Kayaalp и соавт. (1997) используются всего три номенклатуры: 1) ФКУ; 2) не-ФКУ гиперфенилаланинемия (ГФА); 3) варианты ФКУ
По P. Guldberg и соавт. (1998), при рассмотрении ФКУ используются четыре номенклатурные рубрики: 1) классическая ФКУ; 2) умеренная ФКУ; 3) легкая (мягкая) ФКУ; 4) легкая (мягкая) гиперфенилаланинемия (ГФА).
ГФА — основной лабораторный признак болезни, не являющийся эквивалентом ФКУ. ГФА бывает транзиторной и доброкачественной, фиксируется, начиная с достижения фенилаланином в крови уровня >2 мг% (120 мкмоль/л). Все ГФА на уровне свыше 8 мг% (480 мкмоль/л) расцениваются как ФКУ
В практических целях в медико-генетических центрах РФ используется условная классификация ФКУ, основанная на уровнях содержания фенилаланина в сыворотке крови: классическая (тяжелая или типичная) ФКУ — уровень фенилаланина выше 20 мг% (1200 мкмоль/л); 2) средняя ФКУ — 10,1-20,0 мг% (600-1200 мкмоль/л), а также уровень фенилаланина 8,1-10,0 мг%, если он устойчив на фоне физиологической нормы потребления белка в рационе питания; 3) легкая (мягкая) ФКУ (гиперфенилаланинемия, не требующая лечения) — уровень фенилаланина до 8 мг% (480 мкмоль/л).
В современных руководствах разновидности болезни рассматриваются под порядковыми (римскими) цифрами: I (ФКУ классическая/тяжелая), II (ФКУ атипичная — дефицит тетрагидро-биоптерина) или III (ФКУ атипичная — дефицит 6- пирувоилтетрагидроптеринсинтазы); кроме того, выделяют примаптеринурию (ФКУ атипичная) и материнскую ФКУ
Клинические проявления и диагностика фенилкетонурии
При рождении дети с классической фенилкетонурии I выглядят здоровыми, хотя чаще имеют специфический габитус (светлые волосы, голубые глаза, суховатую кожу, иногда с признаками экземы).
При отсутствии своевременного выявления и лечения болезни в течение первых 2 мес жизни у них отмечается появление частой и интенсивной рвоты, повышенной возбудимости, а между 4-м и 9-м месяцами выраженное отставание в психомоторном развитии становится очевидным.
Пациентов отличает специфический («мышиный») запах кожных покровов. Судорожные приступы при ФКУ считаются не характерными для неонатального периода и обычно дебютируют в возрасте до 18 мес (могут исчезать спонтанно).
Из диагностических методов (помимо определения содержания в крови уровней фенила-ланина и тирозина) используются традиционная проба Феллинга, тест Гатри, хроматография, флуориметрия, поиск мутантного гена.
Широко применяются ЭЭГ- и МРТ-исследования. При ЭЭГ-исследовании могут выявляться нарушения (паттерн гипсартимии даже при отсутствии приступов, единичные и множественные фокусы спайк- и полиспайк-разрядов). Данные МРТ-исследования обычно аномальны вне зависимости от проведения/отсутствия лечения ФКУ: на Т2-взвешенном изображении повышение интенсивности сигнала в перивентрикулярном и субкортикальном белом веществе задних отделов гемисфер. Описанные изменения при МРТ-исследовании зависят от уровня содержания фенилаланина в крови.
При ФКУ II клиническая симптоматика отсутствует в периоде новорожденности (появляется в начале 2-го года жизни). Диетотерапия малоэффективна и нередко к 2-3 годам наступает летальный исход.
Клиническая проявления и время дебюта ФКУ III напоминают таковые при ФКУ II.
Профилактика фенилкетонурии
Своевременное выявление фенилкетонурии при использовании соответствующих скрининг-тестов в родильных домах и генетическое консультирование.
Будущим матерям, страдающим ФКУ, для предотвращения повреждения плода до зачатия рекомендуется строго соблюдать диету с низким содержанием фенилаланина, придерживаться ее на протяжении всей беременности, поддерживая уровень фенилаланина ниже 6 мг% (360 мкмоль/л).
Потомство матерей с легкой ФКУ (ФА <6,6 мг% или <400 мкмоль/л) обычно не страдает, а матерям не требуется диетотерапия.
Лечение фенилкетонурии
В настоящее время интенсивно разрабатываются сразу несколько видов альтернативной терапии ФКУ: метод «больших нейтральных аминокислот» (large neutral amino acids), энзимотерапия фенилаланингидроксилазой (phenylalanine hydroxylase enzyme, РАН) и фени-лаланинаммониалиазой (phenylalanine ammonia lyase, PAL), лечение тетрагидробиоптерином. Отметим, что, к сожалению, тетрагидробиоптерин не предназначен для терапии новорожденных и детей первых 4 лет жизни.
Еще один экспериментальный метод лечения фенилкетонурии — введение гена фенилаланингидроксилазы (ФАГ) в пораженные клетки печени. В РФ указанные методы в настоящее время не применяются.
Диетотерапия фенилкетонурии — основной метод лечения.
- Синдром ригидного младенца (stiff baby syndrome) - причины, диагностика, лечение
- Синдром Шварца-Джампеля - причины, диагностика, лечение
- Врожденная (семейная) дисавтономия (синдром Райли-Дэя) - причины, диагностика, лечение
- Гликогеноз тип IIa (болезнь Помпе) - причины, диагностика, лечение
- Инфантильные периферические нейропатии - причины, диагностика, лечение
- Фенилкетонурия (ФКУ) у новорожденных - причины, диагностика, лечение
- Болезнь мочи с запахом кленового сиропа (БМЗКС, лейциноз) у новорожденных - причины, диагностика, лечение
- Галактоземия у новорожденных - причины, диагностика, лечение
- Гистидинемия у новорожденных - причины, диагностика, лечение
- Тирозинемия у новорожденных - причины, диагностика, лечение