
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Патогенез полиглутаминовых форм аутосомно-доминантных атаксий
Среди всех аутосомно-доминантных атаксий механизм нейродегенерации наиболее детально изучен для тех форм, которые относятся к группе полиглутаминовых болезней СЦА1, СЦА2, СЦАЗ, СЦА6, СЦА7, СЦА17 и ДРПЛА.
Центральным звеном патогенеза данных заболеваний является изменение конформации, затрагивающее удлиненную полиглутаминовую цепь мутантной белковой молекулы (Иллариошкин С.Н.). Показано, что при превышении определенного порогового числа глутаминовых остатков в белке его молекула приобретает склонность к формированию особых «шпилечных» структур, образующих затем высокомолекулярные антипараллельные р-тяжи (b-слои) по механизму полярных «застежек-молний» (Pcrutz М. et аl.).
Образование b-складчатых вторичных белковых структур приводит, в свою очередь, к преципитации полиглутаминсодержащих белков в виде нерастворимых амилоидоподобных полимерных комплексов, токсичных для специфических групп нейронов (Paulson Н.).
Подтверждением ключевой роли конформационных изменений мутантных белков в патогенезе полиглутаминовых болезней явилось обнаружение в ядрах дегенерирующих нейронов (клеток Пуркинье и др.) особых сферических включений, содержащих мутантные полиглутаминсодержащие белки (Koyano S. et al., Paulson Н.).
При одной из форм аутосомно-доминантных атаксий — СЦA6 — эти белковые агрегаты выявляются не только в ядрах, но и в цитоплазме нейронов (Ishikawa К. et al.). Формирование полиглутаминсодержащих включений представляет собой универсальный патологический процесс при данном типе нейродегенерации, поскольку такие включения обнаруживаются также в пораженных нейронах у трансгенных животных (млекопитающих и дрозофилы), а также в изолированных культурах клеток, экспрессирующих встроенные в их геном экспандированные участки генов ATXN1 и ATXN2 (Ikeda H.et al., Warrick J. et al.).
В ответ на экспрессию мутантных полиглутаминсодержащих белков в пораженных клетках включаются естественные защитные механизмы. Первая линия защиты — мобилизация вспомогательных белков-шаперонов Hsp40, Hsp70 и др., способствующих «распрямлению» полиглутаминовых конформеров и их дезагрегации (Cummings С. et al., Paulson Н. et al.).
Вторая линия клеточной защиты — активация связанного с молекулярными шаперонами убиквитин-протеасомного пути, т.е. расщепление аномальных убиквитин-меченных белков ферментами протеасомного комплекса цитоплазмы (Cummings С. et al., Chai Y. et al.). Однако в условиях патологии эффективная деградация мутантных белков в протеасомах невозможна, поскольку длинные полиглутамины формируют энергетически стабильную структуру, препятствующую «расплетению» молекулы и ее вхождению в протеолитическую камеру протеасомы (Paulson Н.).
Таким образом, связывание протеасомных компонентов полиглутаминовыми включениями происходит «вхолостую» и приводит лишь к истощению протеасом в клетке и дальнейшему повышению содержания аномальных конформеров.
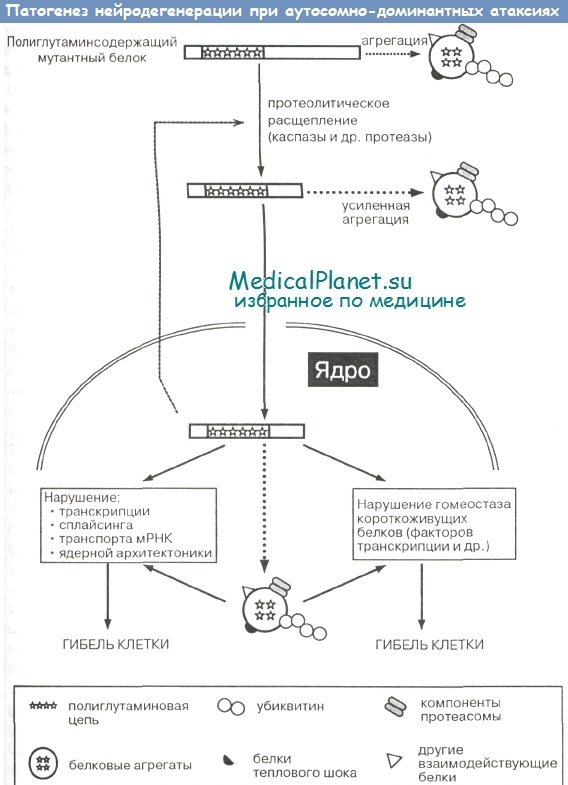
Ключевым фактором нейродегенерации при рассматриваемой группе заболеваний является локализация мутантных полиглутаминсодержащих белков в ядре нейронов (Klement I. et al., Paulson H.). Это может быть связано с тем, что ядро имеет менее совершенную систему дезагрегации и деградации аномальных белков по сравнению с цитоплазмой либо с особенностями внутренней структуры ядра и наличием в нем специфических субдоменов, обладающих способностью концентрировать мутантные белки и стимулировать их агрегацию.
Значение ядерной локализации полиглутаминовых молекул для гибели нейронов подтверждено в эксперименте: экспрессия рекомбинантных мутантных белков с инактивированным ядерным эпитопом (что подавляет экспорт белка в ядро) приводила к значительному уменьшению их токсичности (Кlеment I. et al.). Как установили M.Perez и соавт., при поступлении атаксина-3 в ядро происходит связывание данного белка с ядерным матриксом и изменение его конформации, в результате чего полиглутаминовый эпитоп становится доступным для прямого контакта с другими ядерными белками, формируя чрезвычайно стабильные и нейротоксичные комплексы.
Результаты исследований in vivo и in vitro показывают, что наибольшая степень нейротоксичности полиглутаминов связана не с полноразмерными молекулами, а с их усеченными фрагментами, в составе которых «агрессивный» полиглутаминовый участок максимально открыто экспонирован по отношению к окружающим белкам (Ikeda Н. et al., Paulson Н., Rubinsztein D. et al.). Мутантные продукты полиглутаминовых генов претерпевают расщепление каспазами — особым классом клеточных протсаз, обладающих проапоптозным действием и способностью специфически связываться с полиглутаминами (Wellington С. et al., Sanchez I. et al.).
Активация каспаз приводит к дальнейшему усилению протеолиза и еще более ускоренному образованию наиболее токсичных укороченных полиглутаминсодержащих пептидов, замыкая тем самым своеобразный порочный круг и приводя к неизбежной гибели нейронов по механизму апоптоза. Для полиглутаминовых болезней показано участие и других клеточных протеаз в прогрессирующем расщеплении белка на все более короткие фрагменты (Rubinsztein D. et al.). Расщепление полиглутаминовых белков до более простых пептидов облегчает их проникновение в ядро через ядерную мембрану.
Как установлено в эксперименте, внутриядерные полиглутаминовые включения, будучи важнейшими маркерами нейродегенеративного процесса, могут сами по себе не иметь патологического значения и даже служить своеобразной защитой клетки в ответ на избыток сверхдлинных полиглутаминов — попыткой секвестрировать и связать токсичные пептиды (Sisodia S., Paulson Н., Rubinsztein D. et al.). Каковы же интимные механизмы токсичности непосредственно полиглутаминсодержащих молекул на ранней стадии их экспрессии в клетке и особенно клеточном ядре?
Одним из таких механизмов может быть нарушение транскрипции генов, обусловленное связыванием и инактивацей мутантными белками ряда регуляторных транскрипционных пептидов, в составе которых нередко полиглутаминовые цепи (Willems Р., Nucifora F. et al.). В связи с этим интересно отметить, что продукт гена СЦА17 - белок ТВР (TATA-binding protein) — по своей функции сам является фактором транскрипции (Nakamura К. ct al.).
Кроме того, уже на ранних стадиях патологического процесса мутантные полиглутаминовые цепи посредством формирования перекрестных глутамиллизиновых сшивок могут ковалентно связывать ряд других, лизин-содержащих, ядерных регуляторных пептидов — таких как факторы сплайсинга мРНК, транспортеры мРНК и белков из ядра в цитоплазму и т.п. (Willems Р., Albin R., Tagle D., Kahlem P. et al., Paulson H.).
Результатом этого становится нарушение экспрессии различных генов и распад нормальных механизмов процессинга первичных транскриптов. В пользу такого сценария свидетельствует обнаружение в составе как минимум двух полиглутами-новых белков наследственных атаксий (атаксина-2 и атрофина) аминокислотных мотивов, характерных для РНК-сплайсинговых пептидов (Paulson Н.).
В патогенезе рассматриваемой группы болезней важное значение имеет нарушение взаимодействия мутантных полиглутаминовых цепей с белками-регуляторами апоптоза (каспазами и др.), что на определенном этапе приводит к запуску процессов программируемой гибели нейронов (Housman D.). Индукция апоптоза полиглутаминсодержащими белками выявлена, в частности, на примере атаксин-3-ассоциированных моделей нейродегенерации (Ikeda Н. et al.).
Значение апоптоза в патогенезе полиглутаминовых болезней убедительно продемонстрировано в экспериментах на трансгенной линии дрозофилы и культуре клеток нейронов, экспрeссирующих различные фрагменты мутантного гена ATXN3: на этих моделях удалось установить, что экспрессия антиапоптозного гена Р35 (Warrick J. et al.) либо ингибирование проапоптозной пептидазы каспазы-8 (Sanchez I. et al.) блокировали полиглутаминопосредованную гибель нейронов.
S. Igarashi и соавт. на культуре клеток COS-7 показали, что наблюдаемая в результате экспрессии мутантного белка атрофина реакция апоптоза может частично подавляться при использовании ингибиторов трансглутаминазы (цистамин, монодансил-кадавсрин), подтвердив тем самым роль формируемых трансглутаминазами глутамил-лизиновых межмолекулярных сшивок в образовании белковых агрегатов и апоптотичес-кой смерти нейронов.
Еще одним фактором нейротоксичности на ранней стадии патологического процесса является способность удлиненных полиглутаминов с измененной конформацией формировать эктопические ионные каналы в двуслойных фосфолипидных мембранах (Hirakura Y. et al., Monoi Н.). Это может приводить к выраженным флюктуациям мембранного тока и серьезным нарушениям возбудимости нейронов, а также процессов секреции нейротрансмиттеров и активации клеточных сигнальных путей.
Весьма ранним нейрофизиологическим нарушением при данных заболеваниях является активизация NMDA-рецспторов глутамата, сопровождающаяся феноменом эксайтотоксичности и повышением содержания внутриклеточных ионов Са2+ в пораженных нейронах (Hodgson J. et al). Следует добавить, что у трансгенных мышей с экспансией полиглутаминов накопление мутантного белка в синаптических терминалях сопровождается нарушением везикулярного обратного захвата глутамата (Li Н. et al.), что может свидетельствовать о прямом стимулирующем влиянии полигутаминовых белков на развитие эндогенной эксайтотоксичности.
По мере прогрессирования патологического процесса токсичным, по-видимому, становится непосредственное накопление фибриллярных агрегатов в ядре и цитоплазме нейронов вследствие нарастающего масс-эффекта и прогрессирующей дезорганизации нормальной цитоархитектоники клетки. В состав этих агрегатов могут входить не только полиглутаминовые фрагменты мутантных белков, но и другие клеточные белки, формирующие стойкие связи с патологически удлиненными полиглутаминовыми цепями.
Это подтверждается обнаружением специфического взаимодействия полиглутаминсодержащих пептидов с рядом нормальных белков клетки, выполняющих разнообразные функции - кальмодулином, глицеральдегид-3-фосфат-дсгидрогеназой, лейцин-обогащенным кислым ядерным протеином и др., причем интенсивность связывания этих белков напрямую коррелирует с длиной полиглутаминовых цепей (Burke J. et al., Matilla A. et al., Feigin A.). Весьма вероятно, что нарушение функции связываемых указанных белков вносит свой вклад в патогенез рассматриваемой группы болезней. Так, нарушение функции глицеральдегид-3-фосфат-дегидрогеназы закономерно сопровождается дефектом энергетического метаболизма нейронов (усугубляя тем самым дегенеративные изменения в мозге) и индукцией апоптоза (Ishitani R. et al.).
Связывание удлиненными полиглутаминовыми цепями ацетилтрансферазных ферментов ингибирует ацетилирование структурных хромосомных белков — гистонов, что сопровождается нарушением нормальных механизмов регуляции генной экспрессии (Steffan J. et al.).
Таким образом, для полиглутаминовых аутосомно-доминантных спиноцеребеллярных атаксий характерно многообразие и универсальность молекулярных механизмов, посредством которых происходит реализация неблагоприятного эффекта кодирующих (CAG)n-повторов соответствующих мутантных генов.
- Читать "Патогенез спиноцеребеллярных атаксий - СЦА8, СЦА10 и СЦА12"
Оглавление темы "Аутосомно-доминантные атаксии":- Аутосомно-доминантные атаксии - история изучения
- Классификация аутосомно-доминантных атаксий
- Генетика аутосомно-доминантных атаксий
- Эпидемиология аутосомно-доминантных атаксий - распространенность, частота
- Патогенез полиглутаминовых форм аутосомно-доминантных атаксий
- Патогенез спиноцеребеллярных атаксий - СЦА8, СЦА10 и СЦА12
- Трансгенные модели аутосомно-доминантных атаксий
- Клиника аутосомно-доминантных атаксий и их течение
- Пример аутосомно-доминантной атаксии
- Патоморфология аутосомно-доминантной атаксии - гистология