
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Мукополисахаридозы (МПС) у детей - причины, типы
Под термином «мукополисахаридозы» (МПС) объединяется ряд патологических процессов, в основе которых лежат наследственные нарушения обмена веществ соединительной ткани. Хотя первые описания этих заболеваний относятся к 1917 (Hunter) и 1919 (Hurler) годам, характер биохимических расстройств при них стал известен только через 50 лет, когда Dorfraan, Lorenz, Meyers и соавт. установили, что в моче больных содержится большое количество кислых мукополисахаридов.
В последующем эти заболевания были отнесены в группу генетически детерминированных энзимопатий, при которых в результате неполного распада сульфатированных ГАГ в лизосомах происходит патологическое накопление мукополисахаридов (Dorfman и соавт.). Детальное изучение многими исследователями патогенеза МПС позволило в 1965 году больных этой группы разделить на 6 подгрупп: синдром Гурлер, синдром Гунтера, синдром Санфилиппо, синдром Моркио, синдром Шейе и синдром Марото — Лами.
Для всех заболеваний, относящихся к этой группе, общим является нарушение метаболизма кислых гликозаминогликанов, которое приводит к отложению патологических продуктов обмена веществ в соединительной ткани, прежде всего в костно-хрящевом аппарате с явными его изменениями, а также в отдельных органах и системах (печень, селезенка, роговица глаза, центральная нервная система). Наряду с характерной клинической картиной гипергликозаминогликанурия является наиболее постоянным симптомом заболевания, обнаружение которого имеет решающее значение для постановки диагноза.
Из других присущих МПС особенностей следует отметить наличие метахроматических грануляций в лейкоцитах крови и клетках костного могза (гранулы Рейлли).
Кислые ГАГ, в норме экскретируемые с мочой, представляют собой смесь хондроитинсульфатов А и С, которые подобно гиалуроновой кислоте расщепляются тестикулярной гиалуронидазой. При МПС основными видами кислых ГАГ, выделяющихся с мочой в избытке в разных соотношениях, зависящих от типа заболевания, являются гепарансульфат (ГС), дерматансульфат (ДС) и кератансульфат (КС). Все они устойчивы к гиалуронидазному гидролизу и находятся в моче здоровых лишь в минимальных количествах. При всех типах МПС экскреция кислых ГАГ увеличена в десятки раз по сравнению с нормой.
Больные с разными типами МПС фенотипически очень сходны между собой, так как в их клинической картине наблюдаются сочетания грубых повреждений ряда органов и систем: скелетные деформации, недостаточность умственного развития, нарушение зрения и слуха и др. Уже при первом осмотре врач обращает внимание на характерные внешние признаки больного: диспропорциональный рост, «башенная» форма черепа, грубые черты лица с широкими надбровными дугами и толстыми губами, большой язык, короткая и толстая шея, короткое туловище, кифоз грудного или поясничного отдела позвоночника, большой живот, пупочные и паховые грыжи, короткопалость, тугоподвижность суставов и пр.
Идентификация конкретных типов МПС у детей представляет определенные трудности. Объясняются они прежде всего фенотипическим сходством больных. Тем не менее, в диагностике большое значение придается не только характеру изменений кислых ГАГ и их фракций в моче, но и особенностям клинических проявлений. Для дифференциации МПС различных типов предложена таблица, составленная на основе литературных данных (McKusick).
В последние 3—4 года в изучении генеза МПС была сделана серия открытий, которые повзолили отнести эти заболевания к группе генетически обусловленных энзимопатий. Neufeld и Barton, анализируя ход научных изысканий по этой проблеме, отмечают, что остается только удивляться, почему так долго эти заболевания не были открыты.
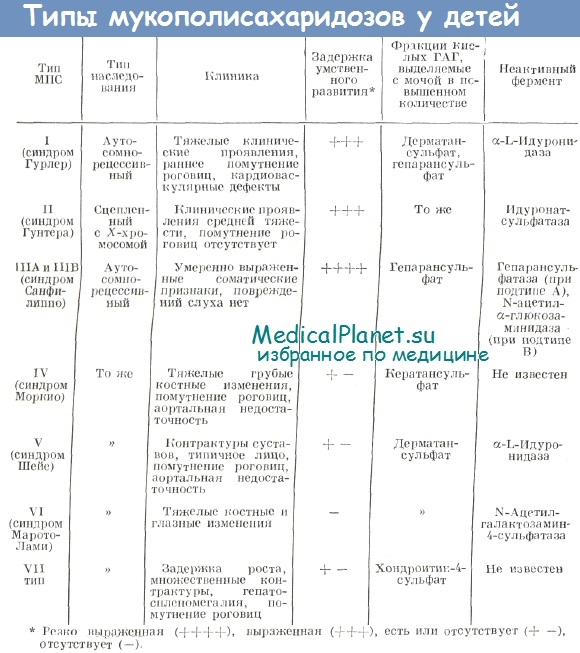
За исключением синдрома Моркио, при всех типах МПС в основе патологического процесса лежат нарушения лизосомального катаболизма полимерных субстанций — дерматансульфата, гепарансульфата и хондроитин-4-сульфата. Распад этих полимеров происходит с помощью различных лизосомальиых ферментов. Если один из них функционально неадекватен, имеется его недостаточность или же он отсутствует вовсе, то последовательность катаболизма нарушается.
В норме распад дерматансульфата и гепарансульфата приводит к образованию сульфата, который экскретируется с мочой, и гексозамина и уроновых кислот, которые вновь вступают в обменный цикл по гликолитическому пути. У больных МПС изменен распад кислых ГАГ и происходит накопление их в лизосомах, что приводит к грубой клеточной патологии и возникновению специфической клинической картины.
Нарушение процесса распада мукополисахаридов наблюдали в 1964 году Van Hoof и Hers, которые при электронном микроскопировании смогли отметить большие вакуоли, наполненные мукополисахаридами у больного с синдромом Гурлер. Проводя аналогию с накоплением гликогена в лизосомах при болезни Помпе, авторы высказали предположение о вторичности аналогичного дефекта при синдроме Гурлер. Эта гипотеза вскоре была экспериментально подтверждена Danes и Beam с помощью культуры клеток. Они доказали, что фибробласты кожи больных с синдромом Гурлер и больных с синдромом Гунтера имели накопления мукополисахаридов.
В последующем Fratantoni и соавт., использовав 35SO4- как маркер для сульфатированных муполисахаридов, показали различие в кинетике мукополисахаридов фибробластов из тканей здоровых и больных с синдромом Гурлер и с синдромом Гунтера. Они высказали мнение, что этот факт не может быть объяснен гиперпродукцией или же, наоборот, снижением секреции мукополисахаридов в клетке больных. Вероятное всего, это связано со сниженной деградацией мукополисахаридов.
Далее был получен очень важный факт, который можно по существу считать ключом к изучавшейся проблеме. Fratantoni и соавт. доказали, что катаболизм мукополисахаридов нормализуется при смешении двух типов, мало отличаемых по внешнему виду, фибробластов больных с синдромом Гурлер и больных с синдромом Гунтера. Среда, предварительно инкубированная с фибробластами больного с синдромом Гунтера, ускоряла катаболизм мукополисахаридов фибробластов больного с синдромом Гурлер и наоборот.
Очень существенно то, что катаболизм Гунтер-клеток не корригировался добавлением среды, предварительно инкубированной с нормальными клетками или же с клетками других генотипов МПС. Это привело к мысли о том, что измененный катаболизм мукополисахаридов в Гунтер-клетках, видимо обусловлен недостатком какого-то вещества, необходимого для катаболизма мукополисахаридов, которое продуцируется Гурлер-клетками и введение которого экзогенно в Гунтер-клетки вызывает корригирующий эффект. Это явление получило в литературе название «взаимная метаболическая кооперация» (Кпарр), а само вещество, обусловливающее этот феномен при синдроме Гунтера,— «корригирующий фактор Гунтера».
Последующие изыскания оказались весьма плодотворными и привели к выявлению аналогичных корригирующих факторов и при других формах МПС. Удалось установить, что в клетках больных с синдромом Гурлер имеется дефицит фактора, специфичного для данного заболевания. Причем этот фактор отличается от аналогичных факторов при синдромах Гунтера, Санфилиппо и Марото — Лами.
Кроме того, была доказана гетерогенность синдрома Санфилиппо, что позволило выделить два генетически разнородных заболевания, обозначенные синдромом Санфилиппо А и синдромом Санфилиппо В, для каждого из которых характерен свой корригирующий фактор. Уникальной корригирующий фактор был найден и при синдроме Марото — Лами. Изучение химической структуры обнаруженных факторов позволило связать их с протеинами. Отсюда было сделано предположение, что успех коррекции объясняется возмещением недостаточности ферментов при этих заболеваниях (Porter и соавт.).
Таким образом, корригирующие факторы стали рассматриваться как кандидаты соответствующих ферментов. Применение радиоизотопных меток 35SО4, 3Н-глюкозамина и 3Н-галактозы позволило выяснить, что особый вид сульфатной группы, который служит субстратом для корригирующего фактора Гунтера, связан с L-идуроновой кислотой. Было показано, что в фибробластах больного с синдромом Гунтера имеется специфическая недостаточность идуронатсульфатазы. Ферментный дефект при синдроме Гурлер установлен одновременно в трех лабораториях, применявших при этом различные методы.
Исследователи доказали, что при этом заболевании имеется дефицит a-L-идуронидазы. Гетерозиготные носители имели промежуточный между больными и здоровыми уровень этого фермента в фибробластах (Bach и соавт., Malaton Dorfman). При синдроме Шейе также обнаружен дефицит a-L-идуронидазы. Установление характера ферментного дефекта при синдроме Санфилиппо также произошло одновременно в нескольких лабораториях. Оказалось, что при синдроме Санфилиппо А снижена активность генарансульфатазы, а при синдроме Сапфилиппо В—N-ацетил-а-глюкозаминидазы (O'Brien). Таким образом, в настоящее время известен характер ферментного дефекта для 5 из 7 типов МПС.
- Рекомендуем далее ознакомиться со статьей "Мукополисахаридоз I типа (синдром Гурлер) у детей - причины, клиника"
Оглавление темы "Заболевания соединительной ткани у детей":- Болезни соединительной ткани у детей
- Мукополисахаридозы (МПС) у детей - причины, типы
- Мукополисахаридоз I типа (синдром Гурлер) у детей - причины, клиника
- Мукополисахаридоз II типа (синдром Гунтера) у детей - причины, клиника
- Мукополисахаридоз III типа (синдром Санфилиппо) у детей - причины, клиника
- Мукополисахаридоз IV типа (синдром Моркио) у детей - причины, клиника
- Мукополисахаридоз V типа (синдром Шейе) у детей - причины, клиника
- Мукополисахаридоз VI типа (синдром Марото—Лами) у детей - причины, клиника
- Болезнь Марфана у детей - причины, клиника
- Дифференциальная диагностика болезни Марфана