
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Врожденный дискератоз (ВДК, синдром Циннсера-Энгмана-Коула)
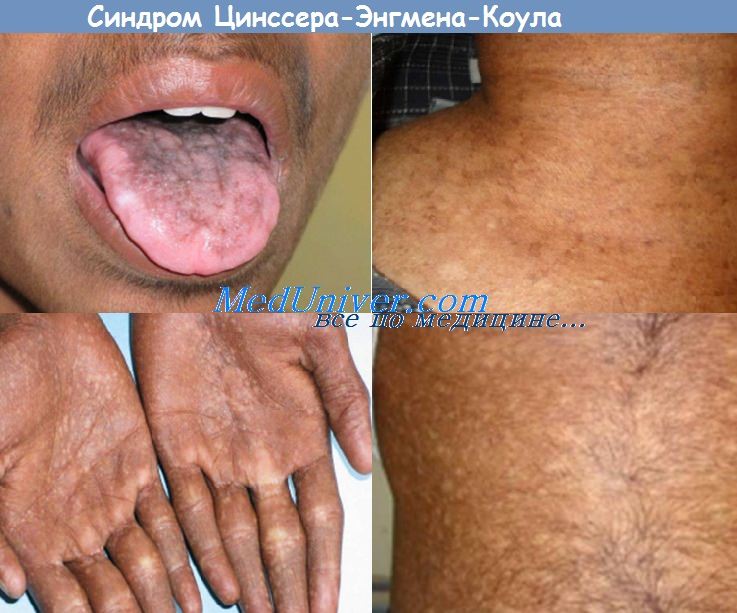
Врожденный дискератоз (ВДК) или синдром Циннсера-Энгмана-Коула характеризуется сетчатой пигментацией кожи, атрофией ногтей, лейкоплакией и недостаточностью функции костного мозга. Дефицит функции костного мозга и злокачественное заболевание развиваются во второй и третьей декадах жизни. Во всех описанных случаях ВДК причинные мутации находятся в компонентах комплекса теломеразы.
Быстро делящиеся соматические клетки экспрессируют низкие, но определяемые уровни активности теломеразы, которая замедляет постоянное укорочение теломеров, происходящее с каждым циклом репликации ДНК и приводящее в итоге к старению клеток (перманентной утрате пролиферативной способности).
Сейчас считается, что врожденный дискератоз (ВДК) развивается вследствие недостаточного поддержания уровня теломеразы, что ограничивает пролиферативную способность гематопоэтических и эпителиальных клеток. Сейчас известно, что в стареющих меланоцитах происходит усиленный синтез меланина, чем, вероятно, объясняется пигментный фенотип ВДК; а критически короткие теломеры могут вынудить клетки войти в состояние «кризиса репликации», в момент которого активация альтернативного механизма «АКТ» удлинения теломеров в отсутствие теломеразы может привести к развитию злокачественных опухолей. Наличие укороченных теломеров при ВДК явилось фактически первым свидетельством роли теломеров в клеточной биологии (старении клеток).
Х-слепленный врожденный дискератоз (ВДК) вызывается мутациями в гене DKC1, расположенном на Xq28, кодирующей дискерин. Женщины-носительницы одной мутантной аллели защищены экспрессией нормальной теломеразы на непораженной аллели. При аутосомно-доминантном ВДК большинство случаев развивается вследствие мутаций в TERC, РНК-компоненте комплекса теломеразы.
TERT (обратная транскриптаза теломеразы) при аутосомно-доминантном ВДК поражается намного реже. Аутосомно-доминантная форма имеет лучший прогноз, вероятно потому, что некоторая активность теломеразы сохраняется благодаря присутствию непораженной аллели.
В аутосомно-рецессивной форме врожденного дискератоза (ВДК) участвуют мутации в ассоциированных с теломеразой белках, таких как NOPIO, NHP2 и TINF2 . Биопсия гиперпигментированной кожи показывает неспецифические изменения, в том числе атрофию эпидермиса, хронический воспалительный инфильтрат с многочисленными меланофагами в верхнем слое дермы. ВДК можно ошибочно принять за синдром Фанкони, для которого характерна карликовость, гипоплазия или аплазия больших пальцев кисти и уменьшение числа костей запястья.
В этом случае пятнистая гиперпигментация туловища, шеи, паховой и подмышечных областей проявляется раньше, чем при ВДК, то есть в первые годы жизни.
Сведения о синдроме Негели-Франческетти-Ядассона, пигментной сетчатой дерматопатии (dermatopathia pigmentosa reticularis), синдроме Даулинга-Дегоса, болезни Китамуры, ретикулярной акропигментации и синдроме Партингтона приведены в отдельных статьях на сайте - просьба пользоваться формой поиска выше.
- Вернуться в оглавление раздела "дерматология."
Оглавление темы "Гипомеланозы и гипермеланозы.":- Виды врожденных гипомеланозов - белых пятен кожи
- Виды приобретенных гипомеланозов - белых пятен кожи
- Виды приобретенных диффузных гипопигментаций - белых пятен на коже
- Виды приобретенных диффузных сосудистых белых пятен на коже (гипопигментации)
- Врожденный диффузный линейный гипермеланоз
- Врожденный дискератоз (ВДК, синдром Циннсера-Энгмана-Коула)