
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизм мутации генов и их полиморфизм
В человеческом геноме генетический код двух здоровых индивидов может иметь множество различий в последовательности ДНК, которые не связаны с заболеваниями или фенотипическими особенностями. Такие изменения внутри нормальной популяции определяются как полиморфизмы.
В действительности, даже в кодирующем участке генома клинически незначимая замена одной пары нуклеотидов, известная как единичный нуклеотидный полиморфизм (SNP), является частым событием и происходит примерно один раз за 250 н.п. Часто однонуклеотидные замены не меняют аминокислотную последовательность; например, замена Ц на Т в третьей позиции пролинового кодона (ЦЦЦ на ЦЦТ) также кодирует пролин и относится к молчащим мутациям. Некоторые однонуклеотидные замены, тем не менее, меняют природу аминокислоты; например, замена Ц на Г во второй позиции того же пролинового кодона (ЦЦЦ на ЦГЦ) меняет пролин на аргинин.
Далее становится важно определить, является ли миссенс-замена непатогенным полиморфизмом или патогенной мутацией, как это представлено выше. Факторами, способствующими формированию патогенной мутации, являются: выделение последовательности только у фенотипически больных людей в определенной семье, аминокислотная замена в эволюционно консервативном участке, замена, воздействующая на функцию белка (размер, заряд, конформация и т.д.), нуклеотидное переключение, которое не обнаружено по крайней мере в 100 контрольных хромосомах, выбранных по этническому признаку. Непатогенный полиморфизм не всегда включает в себя только однонуклеотидные замены; иногда делеции или вставки могут также быть непатогенными.
Мутация может быть определена как замена в химическом составе гена. Миссенс-мутация — это замена одной аминокислоты на другую. Мутациями также являются вставки или делеции оснований, последствия которых зависят от того, сдвинут они нормальную рамку считывания или нет, так же как для нонсенс-мутаций, что может привести к преждевременной терминации трансляции. Например, делеция одного нуклеотида внутри экзона вызывает сдвиг рамки считывания, что обычно приводит к преждевременному стоп-кодону и в итоге дает укороченный белок или нестабильную мРНК, которая быстро разрушается в клетке.
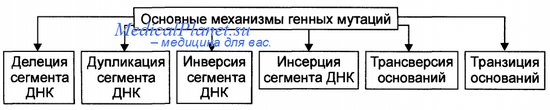
Тем не менее деления трех нуклеотидов (или количества, кратного трем) не будет значительно нарушать полную рамку считывания, и последствия будут зависеть от природы нуклеотидов, которые выпали. Нонсенс мутации обычно, но не всегда, случаются в CpG динуклеотидах, где часто происходит метилирование цитозина. Наследственная химическая нестабильность такого модифицированного цитозина приводит к высокому уровню его превращения в тимин. Изменение кодона (например, из ЦГА в ТГА), приведет к замене остатка аргинина на стоп-кодон. Нонсенс-мутации обычно приводят к снижению или отсутствию экспрессии мутантного аллеля на уровне мРНК или белка.
В гетерозиготном состоянии это может клинически не проявляться [например, родители больных узловым буллезным эпидермолизом Херлитца являются обычно носителями нонсенс-мутаций в одном из генов ламинина-332 или ламинина-5, но сами не имеют кожных проявлений], однако гетерозиготная нонсенс-мутация в гене десмоплакина может, например, вызвать такое аутосомно-доминантное кожное заболевание, как стриарная ладонно-подошвенная кератодерма. Этот феномен называется гаплонедостаточностью (т.е. половина от нормального количества белка недостаточна для поддержания его нормальной функции).
В отличие от изменений кодирующей последовательности, которые вызывают сдвиг рамки считывания, миссенс- или нонсенс-мутаций, приблизительно 15% всех мутаций включают в себя изменения в последовательности гена близко на границе между нитроном и экзоном, и известны как мутации сайта сплайсинга.
Этот тип мутаций может разрушить акцепторный и донорский участки сайта сплайсинга, которые в норме вырезают нитроны во время транскрипции гена. Последствия мутации сайта сплайсинга комплексные; иногда они ведут к пропуску соседнего экзона, а иногда вызывают образование новых мРНК транскриптов, используя скрытые сайты сплайсинга в соседнем экзоне или интроне. Мутации в одном гене не всегда ведут к одному наследственному заболеванию. Например, мутации в гене ERCC2 могут привести к пигментной ксеродерме (типа D), трихотиодистрофии или к цереброфацио-скелетному синдрому, в зависимости от расположения и типа мутации.
Другие транс-активные факторы могут изменять фенотипическую экспрессию дальше. Эта ситуация известна как гетерогенность аллелей. Наоборот, некоторые наследственные заболевания вызваны мутациями более чем одного гена, например узловой буллезный эпидермозиз не-Херлитца может быть вызван мутациями в генах COL17A1, LAMA3, LAMB3 или LAMBC2. Этот феномен называется генетической гетерогенностью. Кроме того, одна и та же мутация в определенном гене может привести к различной клинической тяжести заболевания. Вариабельность фенотипа при данном генотипе определяется как "экспрессивность". Если у человека с таким генотипом нет фенотипических проявлений, болезнь является непенетрантной. Вариабельность в экспрессии отражает комплексные взаимодействия между мутацией, модифицирующими генами, эпигенетическими факторами и окружающей средой и показывает, что объяснение/ интерпретация того, что делает специфичная мутация гена включает больше, чем просто нахождение одного участка мутировавшей ДНК в единичном гене.
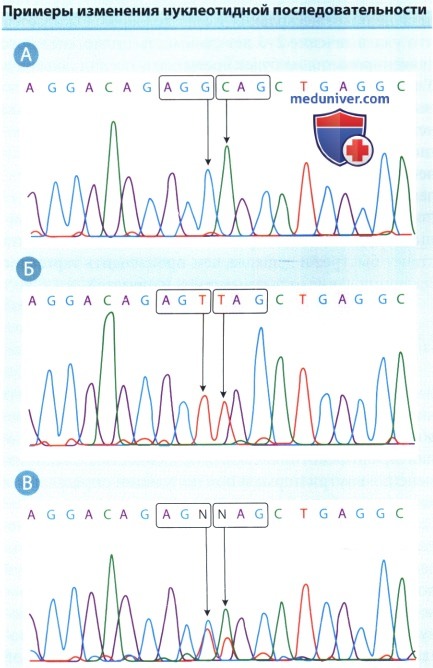
А. Выделены два смежных кодона. Кодон ААГ, обведенный красным, кодирует аргинин, а кодон в синем квадрате, ЦАГ, кодирует глутамин.
Б. При сиквенсе показана гомозиготная замена двух нуклеотидов. В красном квадрате ААГ читается как АГТ (т.е. кодирует серин вместо аргинина).
Это распространенный вариант последовательности в нормальной популяции, и он обозначается как непатогенный миссенс-полиморфизм.
Напротив, в синем квадрате кодон глутамина ЦАГ читается как ТАГ, который является стоп-кодоном. Это пример гомозиготной нонсенс-мутации.
В. Это последовательность от одного из родителей человека, у которого выявлена последовательность Б, она показывает гетеро-зиготность обоих вариантов:
миссенс-полиморфизма АГГ >АГТ и нонсенс мутации ЦАГ>ТАГ, определяя, что пробанд является носителем обоих изменений последовательности.
- Рекомендуем далее ознакомиться со статьей "Менделевские болезни и варианты их наследования"
Оглавление темы "Генетика заболеваний кожи.":- Значение изучения генома человека в дерматологии
- Структура хромосомы и генов человека
- Механизмы и этапы экспрессии генов
- Выявление генов ответственных за заболевание
- Механизм мутации генов и их полиморфизм
- Менделевские болезни и варианты их наследования
- Хромосомные болезни с кожными проявлениями
- Митохондриальные болезни с кожными проявлениями
- Комплексная генетика кожных болезней
- Мозаицизм в генетике кожных болезней