
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Кожа при фенилкетонурии (ФКУ) - диагностика, лечение
Фенилкетонурия - краткий обзор:
- Аутосомно-рецессивное заболевание, вызванное дефицитом фенилалнингидроксилазы (интернет-вариант базы данных OMIM (Online Mendelian Inheritance in Man) № 261600). Ген находится на участке хромосомы 12q22—24.1; частота встречаемости один на 10000 живых новорожденных среди европейцев.
- К клиническим симптомам относятся задержка умственного развития, генерализованная гипопигментация, светлые волосы, голубые глаза, экзематозная сыпь, рвота в грудном возрасте, гиперактивность и судороги, мышиный запах.
- Диагноз подтверждается при положительном результате анализа мочи на хлорид железа.
- Лечение заключается в исключении фенилаланина из диеты.
- Дифференциальная диагностика проводится с альбинизмом, синдромом Чедиака-Хигаси и склеродермой.
- Возможна пренатальная диагностика путем получения образцов хорионических ворсинок/амниоцентеза, измерения уровня метаболитов и анализа ДНК.
Фенилкетонурия (фенилпируватная олигофрения, ФКУ) —это аутосомно-рецессивное нарушение метаболизма ароматической аминокислоты (OMIM #261600), при котором фенилаланин не может превращаться в тирозин. Открытие фенилкетонурии более 70 лет назад позволило установить связь между метаболическим заболеванием и умственной отсталостью, и привело к всемирному распространению программ неонатального скрининга. К тому же, стало очевидно, что эффективное лечение может привести к практически полному излечению этих пациентов.
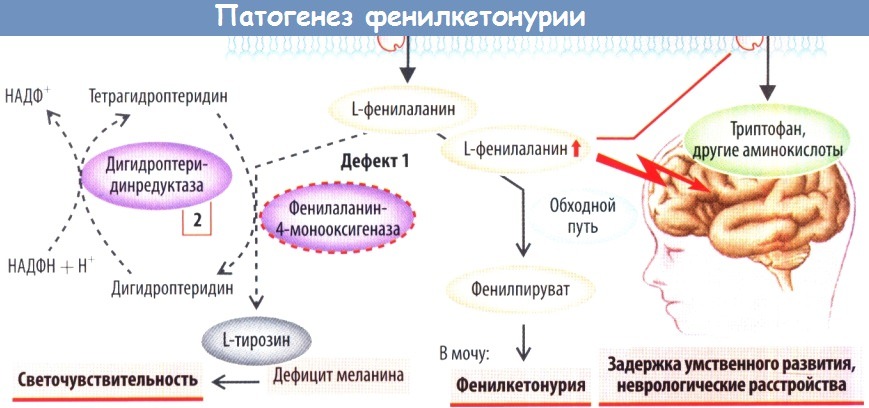
Причиной фенилкетонурии является дефицит печеночной фенилаланингидроксилазы или кофакторов этого фермента. Недостаточность фенилаланин гидроксилазы — это, по всей видимости, болезнь аномального фолдинга белков, при которой глобальные конформационные нарушения препятствуют необходимому для физиологичного функционирования ферментов молекулярному взаимодействию. Существуют дополнительные причины, которые могут приводить к гиперфенилаланинемии; было выделено восемь подтипов заболевания. К гиперфенилаланинемии может приводить материнская ФКУ. Если мать не придерживается специальной диеты, у ребенка имеется риск развития типичных симптомов ФКУ. Дефекты метаболизма биоптерина, преждевременные роды, болезни печени, применение таких препаратов, как меотрексат, а также диеты с высоким содержанием белка могут вызывать развитие симптомов, схожих с симптомами ФКУ. В настоящее время у большинства новорожденных с гиперфенилаланинемей возможно проведение основанного на исследовании генотипа прогноза и классификации биохимического фенотипа.
а) Эпидемиология. Частота встречаемости фенилкетонурии (ФКУ) сильно различается в разных странах. Наибольшая распространенность отмечается в Ирландии и Шотландии (1:4000), в то время как в Финляндии болеет лишь один ребенок на 40000 новорожденных. Общая частота встречаемости в странах северной Европы составляет почти один на 10000 новорожденных, в США в популяции белых людей частота классической ФКУ была оценена как один на 11000 новорожденных. Среди чернокожего населения частота этого заболевания составляет лишь около одного на 50000 новорожденных.
б) Клиника фенилкетонурии (ФКУ). Отличительными клиническими признаками фенилкетонурии являются задержка умственного развития, диффузная гипопигментация, судороги, экзематозный дерматит и фоточувствительность. Дети рождаются без признаков болезни, первые клинические психомоторные симптомы появляются на 4-24 месяце жизни. К ранним симптомам относится выраженная рвота. Наиболее важными нарушениями являются выраженная задержка умственного развития с беспокойством, гиперактивностью, самодеструктивным поведением, гипертонией и судорогами. Отмечаются специфичная походка и повышенные сухожильные рефлексы. Довольно часто наблюдаются микроцефалия, кальцификация головного мозга и катаракты.
Характерен мышиный запах мочи и пота, вызванный фенилаланиновой кислотой.
Ранняя инфантильная экзема, неотличимая от атопического дерматита, может быть одним из первых признаков фенилкетонурии и в течение первого года жизни встречается у 20-50% больных детей. Позднее, частота этого признака может быть даже выше. У некоторых пациентов сообщалось о значительном улучшении проявлений экземы и себорейного дерматита при диете с низким содержанием фенилаланина; также предполагается возможная роль переработки измененного биотина. Следующим ключевым кожным симптомом при фенилкетонурии является распад пигмента, при этом у 90% пациентов отмечается выразительная бледная пигментация, светлые волосы и голубые глаза. Этот симптом является обратимым при диете с ограниченным содержанием фенилланина. Изменения цвета могут быть особенно заметными у чернокожих (редко) или пациентов с ФКУ из Японии, чей цвет глаз или волос может очень сильно отличаться от других представителей этой этнической группы.
Повышенное содержание фенилаланина и продуктов его оксидации (фенилпируватная кислота, о-гидроксифеилацетовая кислота) ингибируют фермент тирозиназу и, таким образом, снижают меланизацию.
У пациентов развивается умеренная фоточувствительность. Характерна дематозная склеродерма конечностей, не поражающая кисти и стопы. При ФКУ также наблюдались бляшки, каплевидная или генерализованная склеродермия и атрофодермия Пазини-Пьери, а также лишайный склероз и болезнь белых пятен.
в) Анализы для диагностики фенилкетонурии. Диагноз фенилкетонурии основывается на выявлении повышенных уровней фенилаланина в сыворотке крови (20 мг/дл или выше, что в 10-50 раз выше нормального уровня). Уровни тирозина в плазме могут быть нормальными или повышенными. Кроме того, могут выявляться повышенные уровни мочевой фенилпируватной кислоты. К дополнительным лабораторным показателям относятся дефицит фенилаланингидроксилазы, фенилпируватная ацидемия и повышение уровней о-гидроксифеилацетовой кислоты и фенилацетилглутамина. Повышение уровней фенилпируватной кислоты могут определяться при типичном зеленом окрашивании мочи после добавления в нее 10% хлорида железа.
В ряде стран скрининг на ФКУ проводится по неонатальным программам, либо путем полуколичественного метода бактериального ингибирования, при котором при помощи микробиологической процедуры (тест Guthrie) в нескольких каплях капиллярной крови можно выявить повышенный уровень фенилаланина, либо методом количественной химической реакции (тест Quantase). У больных детей уровни фенилаланина сыворотки начинают повышаться на третий или четвертый день жизни. Пренатальная диагностика возможна при амниоцентезе или заборе образцов хорионических ворсинок с идентификацией гена. Преимплантационная генетическая диагностика на ФКУ позволяет исключить рождение детей с этим заболеванием.
г) Патофизиология и история. Классический тип ФКУ вызывается дефицитом фенилаланингидроксилазы или ее кофактора тетрагидробиоптерином с последующим накоплением предшественника фенилаланина. Нормальное соотношение между фенилаланином и тирозином составляет 1:1, в то время как при ФКУ оно достигает более 3:1. Широко распространено мнение, что распад пигмента является результатом ингибирующего эффекта фенилаланина на тирозиназу. В биопсийном материале обнаруживается повышенное число фибробластов и гистиоцитов, а также атрофия придатков кожи. Эластические волокна определяются в небольшом количестве и являются фрагментированными и, таким образом, отличаются от эластических волокон при истинной склеродерме. Гистологическое исследование демонстрирует нормальное число меланоцитов, но сниженное число зрелых меланосом.

д) Дифференциальная диагностика. Атопический дерматит и склеродерма, как первичные заболевания, должны быть дифференцированы от фенилкетонурии (ФКУ). У недоношенных детей и пациентов с болезнями печени также могут выявляться повышенные уровни фенилаланина в плазме крови.
е) Лечение фенилкетонурии (ФКУ). При диете с низким содержанием фенилаланина, которая, вероятно, должна соблюдаться в течение всей жизни, изменения цвета кожи, фоточувствительность, характерный запах и экзема являются обратимыми. Кроме того, раннее соблюдение диеты может значительно уменьшать задержку умственного развития, и если уровни фенилаланина в крови поддерживаются на приемлемых цифрах в раннем детском возрасте, можно ожидать нормального интеллектуального развития или нижней границы его нормы. Так или иначе, строгий контроль за соблюдением диеты может приводить к дефициту белка и экзематозному дерматиту. Недавно выяснилось, что сапроптерин повышает толерантность к фенилаланину в процессе обеспечения контроля уровня фенилаланина в крови.

- Рекомендуем далее ознакомиться со статьей "Кожа при аргининосукциниловой ацидурии - диагностика, лечение"
Редактор: Искандер Милевски. Дата публикации: 29.11.2018