
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Артерио-венозные мальформации кожи: причины, клиника, диагностика, лечение
Артерио-венозные мальформации - краткий обзор:
• Распространенность в мире: редкая, точная частота не установлена.
• Врожденные мальформации с высоким кровотоком, которые могут протекать латентно вплоть до пубертатного периода.
• Гистологически представлены прямыми коммуникациями между артериями и венами.
• Как правило, спорадические, но могут иметь генетическую предрасположенность, как в случае капиллярных мальформаций, артерио-венозных мальформаций или наследственной геморрагической телеангиэктазии.
• Могут входить в состав синдрома Паркса-Вебера или синдрома множественных гамартом (ген PTEN).
• Из всех сосудистых мальформаций представляют наибольшие трудности для терапии; необходим мультидисциплинарный подход.
Кожные сосудистые мальформации с интенсивным кровотоком чаще всего являются АВМ, реже — артерио-венозными фистулами (АВФ). АВФ, как правило, травматичные. АВМ характеризуются наличием «очага», эпицентра поражения с прямыми сообщениями между множественными приносящими артериями и дренажными венами, при отсутствии промежуточного звена нормальных капилляров. АВМ представлены деформированными артериями и венами с утолщенными мышечными стенками вследствие артериовенозного шунтирования и фиброза.
а) Эпидемиология. Частота встречаемости артерио-венозных мальформаций (АВМ) неизвестна. Это редкая, как правило, спорадическая сосудистая мальформация с интенсивным кровотоком. Может поражать любые ткани и органы. В 70% АВМ локализуются в области головы и шеи и наблюдаются уже при рождении, но часто появляются в пубертатном периоде или после травмы. Никогда не регрессируют самостоятельно. Наследственная геморрагическая телеангиэктазия (НГТ) — это аутосомно наследуемое заболевание, распространенность которого составляет 1 случай на 10000 человек.

б) Этиология артерио-венозных мальформаций (АВМ). Этиология АВМ неизвестна. Лежащий в основе предрасполагающий генетический дефект выявлен для сосудистых мальформаций с высоким кровотоком, являющихся частью следующих синдромов — КМ-АВМ (OMIM #608354), НГТ (OMIM #187300) или синдром множественных гамартом (PHTS — PTEN Hamartoma Tumor Syndrome) (OMIM #153480 и #158350). Недавно описанное состояние КМ-АВМ сочетает в себе множественные мелкие атипичные КМ с элементами с интенсивным кровотоком (АВМ или АВФ). Мальформация (КМ-АВМ) наследуется по аутосомно-доминантному типу, пенетрантность составляет примерно 95%. Причиной заболевания являются мутации с полной потерей функции в гене KASAL. Высокое разнообразие фенотипических проявлений в кругу одной семьи может объясняться сочетанной соматической мутацией. Частота встречаемости КМ-АВМ неизвестна.
Молекулы, участвующие в сигнальном пути трансформирующего фактора роста-β, предрасполагают к образованию АВМ/АВФ, аналогично тому, как мутации с потерей функции в эндоглине и активин-подобной рецепторной тирозинкиназе вызывают наследственную геморрагическую телеангиэктазию в очагах с интенсивным кровотоком в коже, слизистых оболочках и внутренних органах (легких, головном мозге и печени) (ОМIМ #187300).
Причиной АВМ может быть мутация с потерей функции в PTEN, гене-супрессоре опухолевого роста, которая отмечается у большинства пациентов с PTHS (синдромом множественных гамартом и мутацией в PTEN) (OMIM #158350 8с 153480). Это редкое врожденное, аутосомно-доминантное заболевание характеризуется триадой из макроцефалии, множественных липом и артериовенозных мальформаций, часто многоочаговых, ассоциированных с эктопией подкожно-жировой клетчатки и разрушающих архитектонику тканей.
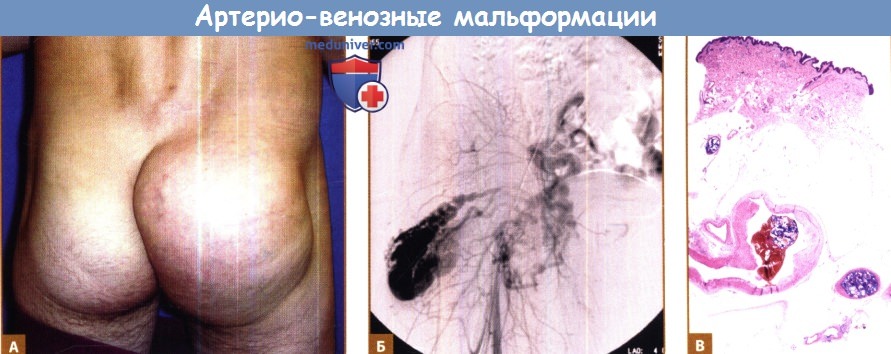
Б. Артериография, демонстрирующая очаг мальформации.
В. Гистологическая картина, видно прямое сообщение между артерией и веной, с тромботической массой, образовавшейся после эмболизации.
в) Клиника артерио-венозных мальформаций (АВМ). В детском возрасте АВМ часто принимают за КМ или гемангиомы, так как при рождении они, как правило, проявляются тусклыми нечеткими пятнами красного цвета. Однако при пальпации в данных патологических элементах можно выявить наличие интенсивного кровотока. Около одной трети АВМ отмечаются при рождении, еще одна треть появляется в детском возрасте или пубертатном периоде, оставшиеся случаи выявляются у взрослых. Провоцирующими факторами роста АВМ обычно являются пубертатный период и травмы. Согласно классификации по Schobinger, выделяется несколько стадий АВМ. АВМ проявляются кожными красно-пурпурными теплыми на ощупь образованиями, сопровождающимися дрожанием, шумом или пульсацией повышенной амплитуды. С течением времени они прогрессируют от I стадии до III и даже IV и никогда самостоятельно не регрессируют.
1. КМ-АВМ. У 18-20% пациентов КМ-АВМ представлены множественными атипичными КМ в сочетании с очагами с интенсивным кровотоком — АВМ, АВФ или синдромом Паркса-Вебера. Выраженность поражения сильно различается в кругу отдельных семей — от мелких бессимптомных КМ до жизнеугрожающих КМ-АВМ. В отличие от спорадических, КМ при КМ-АВМ часто множественные и имеют случайное распространение. Диаметр очагов варьирует от нескольких миллиметров до нескольких сантиметров. Патологические элементы имеют кругловато-овальную форму, розовый, красный или коричневый цвет и иногда узкий бледный венчик. Очаги поражения хорошо отграничены и бледнеют при надавливании под стеклом. Некоторые из них выявляются уже при рождении, другие появляются позднее. Они увеличиваются по мере роста ребенка и не вызывают клинических жалоб. Очаги с интенсивным кровотоком (АВМ или АВФ) локализуются либо на коже, либо в подкожно-жировой клетчатке (с вовлечением или без вовле-ния в патологический процесс мышц и костей) или в головном мозге.
Около 10-15% пациентов с КМ-АВМ страдают синдромом Паркса-Вебера с поражением либо нижней (две трети пациентов), либо верхней конечности (одна треть пациентов). АВМ могут протекать бессимптомно, но в зависимости от их локализации и размера, возникают различные осложнения.
2. Наследственная геморрагическая телеангиэктазия (НГТ). Наследственная геморрагическая телеангиэктазия (НГТ) проявляется множественными телеангиэктазиями на коже и слизистых оболочках в сочетании с висцеральными, легочными и церебральными очагами с интенсивным кровотоком. Частым симптомом является носовое кровотечение, которое может угрожать жизни.
3. Синдром множественных гамартом (ген PTEN). В синдром множественных гамартом PTEN (PHTS) объединены синдром Банаян-Райли-Рувалькаба (СБРР) и синдром Коудена, так как в 60-81% случаев выявляются мутации в гене PTEN. К типичным симптомам относятся макроцефалия, наличие пигментных пятен (по типу веснушек) на половом члене, множественные аномалии развития вен головного мозга, сосудистые мальформации с высоким кровотоком (54%) и повышенный риск злокачественных опухолей. Сосудистые мальформации часто бывают мультиочаговыми (57%) и сочетаются с эктопическим отложением подкожно-жировой клетчатки и нарушением нормальной архитектоники тканей.
4. Синдром Паркса-Вебера. Синдром Паркса-Вебера (OMIM #608355) был впервые описан Ф. Парксом Вебером в 1018 г. Это заболевание характеризуется наличием крупного врожденного кожного сосудистого пятна на одной из конечностей, в сочетании с гипертрофией мягких и скелетных тканей пораженной конечности и множественными подлежащими артерио-венозными микрофистулами.10 Пораженная конечность имеет большую длину и толщину, чем здоровая. С течением времени признаки заболевания прогрессируют. У пациентов, страдающих синдромом Паркса-Вебера, в молодом возрасте может развиваться застойная сердечная недостаточность.
5. Синдром Бонне-Дешама-Бланка или Уайберна-Мейсона. Синдром Бонне-Дешама-Бланка или Уайберна-Мейсона является спорадической синдромной АВМ, расположенной в центрофациальной и/или гемифациальной области, в сочетании с поражением окулоорбитальных и церебральных структур. АВМ присутствует с рождения и с возрастом ухудшается. У пациентов наблюдается эпистаксис, экзофтальм и гемианопия. Возможна также задержка психического развития.
6. Синдром Кобба. Синдром Кобба — еще одна спорадическая синдромная АВМ, объединяющая артериовенозные мальформации кожи и спинного мозга, находящиеся в одном сегменте. Синдром проявляется в детстве в форме неврологических осложнений (боль, сенсорные и моторные нарушения и нейрогенные дисфункции мочевого пузыря), которые зависят от локализации и распространенности АВМ. Лечение состоит в полном хирургическом удалении очага АВМ в спинном мозге, когда это возможно.



г) Визуализирующие методы исследования. При проведении ультразвукового исследования с цветной допплерографией выявляется не какое-либо образование, а скорее зона интенсивного артериального кровотока и пульсирующего венозного кровотока с низким сопротивлением. Сосуды деформированы. При поражении конечностей наблюдение проводится путем оценки артериального оттока от пораженной конечности по сравнению со здоровой. Компьютерную томографию заместила МРТ, которая лучше демонстрирует распространение АВМ и позволяет проводить дифференциальную диагностику с гемангиомами и ВМ. Патогномоничным признаком АВМ являются так называемые «flow voids» — феномен потери сигнала, что соответствует сосудам с высоким кровотоком. До начала лечения необходимо выполнить артериографию для идентификации приносящих питающих артерий и очага поражения.
д) Прогноз и течение артерио-венозных мальформаций (АВМ). АВМ имеют тенденцию к прогрессированию, что приводит к местной деструкции и/или жизнеугрожающему кровотечению. Рост патологического очага провоцируют пубертатный период и травма. Гипертрофия костной ткани является типичным признаком, который вызывает характерную асимметрию лица. АВМ, поражающие конечности, часто приводят к развитию периферической ишемии вследствие синдрома «обкрадывания». Сердечная недостаточность встречается редко (IV стадия по Schobinger), в особенности в детском возрасте. Неадекватное лечение вследствие постановки неверного диагноза или перевязки питающих артерий или частичной резекции очага поражения может иметь драматические последствия и приводить к росту АВМ. КМ при КМ-АВМ безвредны и обычно доставляют лишь косметические проблемы.
е) Лечение артерио-венозных мальформаций (АВМ). Данная сосудистая мальформация с интенсивным кровотоком является наиболее сложной для лечения. Подход к ведению таких больных должен быть мультидисциплинарным. Наблюдение за АВМ должно включать периодическую оценку поражения. При АВМ конечностей или АВФ стабилизировать патологический процесс и предотвратить поражение кожи могут эластические повязки. Целью лечения АВМ является облитерация (при помощи эмболизации) и полное удаление очага поражения. Необходимость раннего вмешательства при «молчащих АВМ» (I стадия) спорно, однако решение данного вопроса должно рассматриваться лишь в том случае, если возможно полное удаление патологического образования.
В отличие от АВФ, суперселективная артериальная эмболизация является лишь паллиативным методом и проводится только при невозможности удаления осложненных АВМ. Эмболизирующие частицы должны достигать эпицентра поражения для предотвращения повторного наполнения очага через новые коллатерали. Другим возможным методом лечения пациентов с предшествующей перевязкой артерий или эмболизацией является непосредственный прокол очага поражения. Хирургическое удаление обычно проводится только после эмболизации с целью уменьшения интраоперационной кровопотери. АВМ требует широкого иссечения. При реконструктивных операциях часто требуется микрохирургическая пересадка свободного лоскута. После любого лечения необходимо наблюдение в течение по меньшей мере пяти лет с ежегодным проведением УЗИс допплерографией и/ или МРТ. Для пациентов с синдромом Паркса-Вебера лечение должно быть как можно более консервативным (например, применение эластических повязок). Для устранения разницы длин нижних конечностей может потребоваться эпифизиодез. Однако эта процедура в некоторых случаях может усугубить течение АВМ.
Для лечения пациентов с НГТ применялись многочисленные методы с целью уменьшения кровоточивости, в частности, местные противовоспалительные препараты, лазеры и хирургические мероприятия. Из-за экстенсивности очагов, все эти мероприятия обеспечивают лишь ограниченные бессимптомные промежутки. Недавно для уменьшения частоты и длительности носовых кровотечений у пациентов с НГТ с успехом применили талидомид.
ж) Список литературы:
- Brouillard Р, Vikkula М: Vascular malformations: Localized defects in vascular morphogenesis. Clin Genet 63(5):340-351, 2003
- Alomari AI: CLOVE(S) syndrome: Expanding the acronym. Am J Med Genet A 149A(2):294; author reply 295, 2009
- Enjolras O, Chapot R, Merland JJ: Vascular anomalies and the growth of limbs: A review. J Pediatr Orthop B13(6):349-357, 2004
- Boon LM, Mulliken JB, Vikkula M: RASA1: Variable phenotype with capillary and arteriovenous malformations. Curr Opin Genet Dev 15(3):265-269, 2005
- Boukobza M et al: [Sturge-Weber syndrome. The current neuroradiologic data], J Radiol 81 (7):765—771, 2000
- Boon LM et al: Glomuvenous malformation (glomangioma) and venous malformation: Distinct clinicopathologic and genetic entities. Arch Dermatol 140(8):971-976, 2004
- Dompmartin A, Vikkula M, Boon LM: Phlebology 25(5): 224-235, 2010
- Wassef M, Enjolras O: [Superficial vascular malformations: Classification and histopathology]. Ann Pathol 19(3):253—264,1999
- Sirvente J et al: Frequency and phenotypes of cutaneous vascular malformations in a consecutive series of 417 patients with familial cerebral cavernous malformations. J Eur Acad Dermatol Venereol 23(9):1066-1072, 2009
- Konez O, Burrows PE: Magnetic resonance of vascular anomalies. Magn P.eson Imaging Clin N Am 10(2):363—388, VII, 2002
- Рекомендуем далее ознакомиться со статьей "Хроническая артериальная недостаточность (ХАН, ПАБ, периферическая артериальная болезнь): этиология, патогенез"
Редактор: Искандер Милевски. Дата публикации: 10.3.2019