
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Мышечная дистрофия Дюшена. Прогрессирующая мышечная дистрофия Бекера.
Заболевание встречается в двух клинических формах, являющихся аллельными генетическими вариантами: прогрессирующей мышечной дистрофии Дюшенна (ПМДД) и Бекера (ПМДБ). Первый вариант был описан еще в 1868 г., его распространенность составляет 1 на 3—3,5 тыс. новорожденных мальчиков. О наличии второго варианта П. Бекер сообщил в i955 г. Он встречается с частотой 1 на 20-25 тыс. лиц мужского пола.
Заболевание возникает в результате мутаций в гене дистрофина, локализованном вобласти Хр21. Приблизительно 30% всех случаев связаны с мутациями de novo в яйцеклетке матери больного, а остальные 70% — обусловлены гетерозиготностью матери по патологической мутации в гене дистрофина. Считается, что 6—7% всех спорадических случаев - результат гонадного мозаицизма (наличия в яичниках женщины нескольких генераций ооцитов с нормальными и мутантными аллеями гена дистрофина). Основной тип мутации, обнаруженный у 60%—70% больных, - это крупные делеции, захватывающие один или несколько экзонов гена и локализующиеся в двух «горячих» регионах - в области 5'-конца (экзоны 6-19) и З'-конца (экзоны 40—43). У 5% больных выявлены дупликации, в остальных случаях — точковые мутации в различных участках гена.
Особенности клинических проявлений при двух аллельных вариантах заболевания связывают с различиями в типах мутаций в гене дистрофина. При ПМДД делеции в гене дистрофина в большинстве случаев приводят к сдвигу рамки считывания и преждевременной терминации трансляции, при этом синтез белка прекращается. При ПМДБ структурные перестройки гена не меняют рамку считывания, ДНК-полимераза может «перескакивать» делегированные экзоны, что приводит к синтезу внутренне усеченного белка, в какой-то мере способного выполнять свои функции.
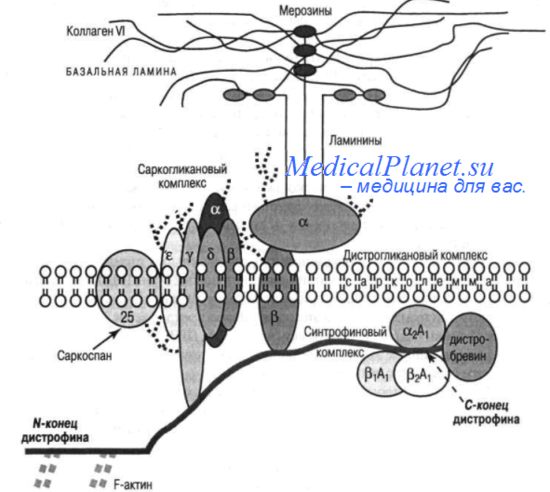
Кодируемый геном белок (дистрофии) относится к семейству спектрин/актиновых мембранных белков цитоскелета и состоит из четырех доменов. Основная функция дистрофина заключается в обеспечении устойчивости и эластичности мышечного волокна при последующих мышечных сокращениях. Это обеспечивается специфической локализацией и строением дистрофина. Располагаясь под сарколеммой мышечного волокна в виде переплетенного антипараллельного димера, дистрофии своим N-концом связан с цитогшазматическим актином, а С-концом - с комплексом мембранных гликопротеинов. Дистрофин-ассоциированный комплекс представляет собой наиболее важный элемент мышечного цитоскелета, который обеспечивает взаимодействие структур экстра- и интрацеллюлярного матрикса, участвует в регуляции уровня кальция в мышце и в передаче импульсов через мембрану мышечного волокна. При отсутствии дистрофина мембрана разрушается, в ней появляются участки некроза. По мере развития болезни мышечное волокно практически полностью разрушается и замещается соединительнотканными структурами, что приводит к псевдогипертрофии мышц - увеличению их объема при утрате или значительном ослаблении функциональных возможностей.
Первые признаки прогрессирующей мышечной дистрофии Дюшенна появляются в возрасте 1-5 лет. Для большинства больных характерна задержка темпов раннего моторного развития. При начале самостоятельной ходьбы (в возрасте старше 14 мес) отмечаются частые падения, двигательная неловкость, бысграя утомляемость. Постепенно походка становится переваливающейся, возникают затруднения при подъеме по лестнице и из положения на корточках. У больных развивается псевдогипертрофия различных мышц, прежде всего икроножных, дельтовидных, четырехглавых и трехглавых, что создает ложное впечатление атлетического телосложения больного. По мере прогрессирования заболевания псевдогипертрофия мышц трансформируется в гипотрофию. Распространение патологического процесса имеет восходящий характер. Первыми поражаются мышцы тазового пояса и проксимальных отделов нижних конечностей, затем мышцы плечевого пояса, спины и проксимальных отделов верхних конечностей. Уже на ранних стадиях болезни снижаются или угасают сухожильные рефлексы. По мере развития патологического процесса в мышцах возникают вторичные деформации позвоночника (усиление лордоза и кифоза, сколиоз), грудной клетки (по типу седловиной и килевидной) и стоп, а также ретракции сухожилий с развитием контрактур в суставах. Еще один характерный признак — кардиомиопатия, которая проявляется симптомами гипертрофии левого желудочка и аритмией. У 25-30% больных диагностируется олигофрения в степени дебильности.
Пациенты сохраняют способность к самостоятельной ходьбе до 10-12 летнего возраста, после чего пользуются инвалидной коляской. Гибель больных наступает от сердечной недостаточности или от интеркуррентных инфекций до 25-летнего возраста.
Прогрессирующая мышечная дистрофия Бекера манифестирует в возрастном интервале от 10 до 20 лет с появления слабости и утомляемости мышц тазового пояса и ног. Ранними симптомами у значительного числа больных бывают болезненные мышечные спазмы. Клинические проявления сходны с таковыми при ПМДД, однако выражены слабее. При ПМДБ, так же как и при ПМДД, в патологический процесс вовлечен миокард. Гипертрофическая или дилятационная кардиомиопатия диагностируется у 50—60% больных. В 40—50% случаев выявляются гипогенитализм и атрофия яичек. Интеллект, как правило, не страдает. Заболевание прогрессирует достаточно медленно и в большинстве случаев приводит к инвалидизации больного не ранее 40-летнего возраста.
Высокую диагностическую значимость при обоих вариантах заболевания имеет активность специфичного для мышечной ткани фермента — креатинфосфокиназы в плазме крови больного. Этот показатель у больных ПМДД в 50-100 раз превышает норму и может быть выявлен до возникновения выраженных клинических признаков.
Для диагностики и дифференциальной диагностики ПМДД/ПМДБ применяются иммуногистохимические методы анализа дистрофина в биогггате мышечного волокна. При использовании антител к различным доменам дистрофина при ПМДД иммунореактивные формы белка, как правило, не выявляются. У больных с ПМДБ наблюдается прерывистое окрашивание мышц при иммунохимическом анализе, что свидетельствует об относительной сохранности отдельных структур цитоскелета.
Одним из основных методов ДНК-диагностики ПМДД/ПМДБ является детекция делеций одного или нескольких экзонов гена дистрофина, которые обусловливают не менее 60% всех случаев заболевания. Учитывая значительную протяженность гена дистрофина, для упрощения поиска делеций вся его кодирующая часть, начиная с 5'-конца, поделена на 9 субфрагментов, размером ) т.п.н., каждый из которых может быть использован в качестве ДНК-зонда. Наиболее часто используется метод мультиплексной амплификации. При ДНК-диагностике применяют набор нуклеотидных последовательностей наиболее часто мутирующих участков гена и кэп-сайта. Использование набора из 18 пар праймеров для амплификации экзонов, расположенных в основном в 3'- и 5'-концах гена дистрофина, позволяет выявить примерно 98% всех крупных делеций. Метод мультиплексной амплификации прост и не требует длительного времени для проведения анализа. Особое достоинство метода-наличие внутреннего положительного контроля почти в каждом образце, так как делеция одновременно всех амплифицируемых участков встречается крайне редко. Пример диагностики делеций в гене дистрофина представлен на рисунке. Если делению зарегистрировать не удается, поиск дупликаций и точковых мутаций в гене, осуществляется крайне редко, что связано со значительной протяженностью гена дистрофина. В этом случае диагноз основывается на использовании данных клинико-генеалогического, биохимического и иммуногистохимического методов обследования больного.
При необходимости проведения пренатальной диагностики и выявления гетерозиготных носительниц поврежденного гена среди родственниц больного используются косвенные методы ДНК-диагностики, принципы которых изложены в статье. В настоящее время клонировано большое количество как внутри-, так и внегенных зондов, позволяющих обнаружить информативный полиморфизм в 99% семей больных с ПМДД/Б. Однако даже при использовании внутригенных маркеров ошибка метода составляет около 5%, что обусловлено возможностью рекомбинации между полиморфным локусом и мутантным участком гена. Наибольшая информативность косвенных методов ДНК-диагностики ПМДД/ПМДБ может быть достигнута при использовании трех зондов, один из которых внутригенный (центральный), а два фланкируют ген с разных сторон. Точность оценки возрастает в случае отсутствия рекомбинации между фланкирующими и центральным маркерами.
- Читать далее "Синдром тестикулярной феминизации. Х-сцепленное доминантное наследование."
Оглавление темы "Нетрадиционные методы наследования.":1. Болезни с нетрадиционным типом наследования. Х-сцепленное рецессивное наследование.
2. Мышечная дистрофия Дюшена. Прогрессирующая мышечная дистрофия Бекера.
3. Синдром тестикулярной феминизации. Х-сцепленное доминантное наследование.
4. Наследственная мото-сенсорная нейропатия Х типа. Признаки мото-сенсорной нейропатии Х типа.
5. Y сцепленный тип наследования. Митохондриальные болезни.
6. Причины митохондриальных болезней. Патогенез митохондриальных болезней.
7. Характеристика митохондриальных болезней. Клиника митохондриальных заболеваний.
8. Биохимические маркеры митохондриальных заболеваний. Синдром Кернс-Сейра.
9. Синдром Melas. Синдром множественных делеций мтДНК.
10. Болезни геномного импритинга. Причины и механизмы развития геномного импритинга.