
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Локусы генов. Генетическое расстояние между двумя локусами.
Скрининг семей на совместное наследование заболевания и аллелей полиморфных маркеров, распределенных по геному, позволяет выявить маркеры, сцепленные с геном заболевания. Генетическое расстояние между двумя локусами рассчитывается, исходя из частоты рекомбинаций между ними на один мейоз. При анализе сцепления важно различать фактическую рекомбинацию маркеров и кроссинговер хромосом, в результате которого рекомбинация может наступить. Лишь нечетное количество кроссинговеров приводит к рекомбинации, то есть к расхождению признаков. Однако с частотой рекомбинаций между локусами не слишком удобно работать, поскольку это неаддитивная величина, которая не может быть больше 0,5. Аддитивной величиной является генетическое расстояние (тар distance), равное среднему количеству кроссинговеров между двумя точками на один мейоз. Генетическое расстояние удобно использовать при построении карт сцепления и выборе маркеров для анализа сцепления.
Учитывая то обстоятельство, что маркер и исследуемый признак могут быть сцеплены не абсолютно, мы должны предполагать существование определенной частоты рекомбинаций между ними (0). С математической точки зрения локусы не сцеплены, если сигма = 0,5 , и сцеплены, если 0 < 0,5. Для количественного анализа сцепления используется величина, называемая Lod-баллом (отангл. logodds -логарифм шансов). Расчет величины Lod-балла проводят исходя из правдоподобия двух событий:
• наличие данного распределения двух признаков в семье F при условии, что их локусы сцеплены (0 < 8 < 1/2).
• наличие данного распределения аллелей двух признаков в семье F при условии, что их локусы не сцеплены (8 = 1/2)
Величина Lod-балла представляет десятичный логарифм отношения правдоподобий данных гипотез и определяется по формуле:
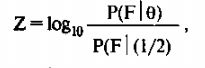
где P(F 18) — правдоподобие первой гипотезы, a P(F|l/2) - правдоподобие второй гипотезы.
По определению,
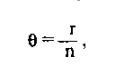
где r — наблюдаемое число рекомбинаций, a n- число информативных мейозов. Правдоподобие каждой из гипотез рассчитывается по формулам:
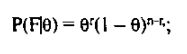
и тогда:
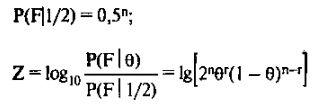
Реально расчет Lod-балла проводится для различных значений 0 с заданным шагом (например, 6 = 0,01; 0,05; 0,10; 0,20; 0,30; 0,40). Значения г и п определяются для каждого маркера из анализа наследования его аллелей в изучаемой семье.
Преимущество использования логарифмов вместо самого отношения правдоподобий состоит в том, что дает возможность суммировать Lod-баллы, полученные для нескольких семей с одним и тем же наследственным заболеванием.
Положительная величина означает, что гипотеза о сцеплении с данным является более вероятной, чем отсутствие сцепления, а отрицательная — наоборот. Из формулы (1) следует, что величина Lod-балла возрастает с увеличением числителя, т.е. вероятности того, что два исследуемых маркера наследуются совместно.
Согласно принципу наибольшего правдоподобия, наилучшей оценкой для реальной частоты рекомбинаций будет 6, при которой достигается максимальная величина Lod-балла. Таким образом, в результате такого рода анализа получаются две величины: 6, характеризующая степень генетического сцепления между маркерами, и Lod, характеризующий определенность, надежность вывода о наличии или отсутствии сцепления. Обычно величина Z > 3 рассматривается как доказательство сцепления (вероятность сцепления между двумя маркерами в 1000 раз больше, чем вероятность их независимой сегрегации). Эта величина выбрана не случайно, и определяется в первую очередь размером генома человека. Упрощенно критерий доказательства сцепления Z >3 можно объяснить следующим образом. Поскольку размер генома составляет 3300 сМ, в нем существует 66 псевдонезависимых локусов, удаленных друг от друга на 50 сМ, и наследуемых независимо. Для достижения 95%-ой достоверности сцепления необходимо иметь двадцатикратный запас по количеству таких точек, т.е. около 1300локусов. Десятичный логарифм данной величины равен 3.2. Таким образом, Z > 3 гарантирует с высокой достоверностью, что обнаруженное сцепление не случайно. При десятикратном увеличении размера генома для доказательства сцепления потребовалась бы величина Z > 4, а при соответствующем уменьшении - достаточна была бы меньшая величина Z . Величина Z < -2 обычно рассматривается как доказательство отсутствия сцепления (отсутствие сцепления в 1 00 раз более вероятно, чем его наличие) и не зависит от размеров изучаемого генома.
Значение сигмы, при которой достигается максимальное значение Lod-балла, может служить оценкой генетического расстояния между двумя сцепленными маркерами. Зависимость между вероятностью рекомбинации 0 и генетическим расстоянием сильно отличается от линейной зависимости при 6 —> 0,5. Для вычисления генетического расстояния между двумя маркерами, исходя из числа наблюдаемых между ними рекомбинаций, используется формула Косамби:
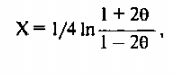
где X - генетическое расстояние в сМ, а сигма- доля рекомбинаций.
При значениях сигма < 0.10 вероятность двойных кроссинговеров между двумя исследуемыми локусами очень мала, и генетическое расстояние можно принять равным рекомбинантной фракции: X = 9.
В настоящее время вычисление Lod-баллов полностью автоматизировано. Программа LINKAGE — пакет компьютерных программ, который дает оценки максимального правдоподобия параметров сцепления на основании всех данных о родословной. Эта программа вычисляет наиболее вероятные гаплотипы членов родословной и использует эти данные для вычисления наиболее вероятной частоты рекомбинации. Программа LINKAGE стала стандартным инструментом при изучении генетического сцепления у человека.
Наряду с изложенной классической схемой геномного скрининга существуют и другие варианты анализа сцепления. Выбор стратеги и поиска генетического сцепления зависит от изучаемого заболевания, от структуры и количества семей, доступных для анализа. Классическая схема полногеномного скрининга имеет ряд ограничений в своем применении. Прежде всего, она плохо применима к генетически гетерогенным заболеваниям. Осуществление картирования такого заболевания, представляет собой серьезную проблему, поскольку сцепление с определенным локусом, которое наблюдается в одних семьях, может отсутствовать в других, приводя к отрицательному суммарному значению Lod-балла и последующему ложному исключению данного локуса из дальнейшего анализа. Один из возможных способов преодоления сложностей картирования, обусловленных генетической гетерогенностью, заключается в выявлении диагностического признака (или группы признаков), по которым различные генетические формы четко отличаются, К сожалению, в большинстве случаев такие клинические признаки выявить не удается. Стандартный подход при проведении картирования локусов генетически гетерогенных заболеваний заключается в выборе семей с четко определенным клиническим фенотипом.
Наилучший подход, позволяющий избежать осложнений, вызванных генетической гетерогенностью, - это работа с одной родословной, достаточно большой для проведения анализа сцепления. Проводя анализ такой родословной с моногенным заболеванием, можно быть уверенным в идентичности генного локуса у больных. Другой подход к эффективному анализу сцепления состоит в изучении отдельной изолированной популяции (этнического, социального, или географического изолята). В этом случае высока вероятностъ того, что мутация, имевшая место у одного из «основателей» популяции, получила распространение в рамках данного изолята и все больные данным заболеванием в популяции являются потомками одного и того же человека.
Еще один способ оптимизации геномного скрининга — анализ сцепления с локусами генов-кандидатов. Геном-кандидатом называют ген, продукт экспрессии которого может прямо или косвенно участвовать в развитии изучаемой болезни. Весьма плодотворным для поиска генов-кандидатов оказался подход, заключающийся в исследовании генов, мутации в которых приводят к сходной патологии у трансгенных животных. Например, ген EGR2 был предложен в качестве гена-кандидата для тяжелых форм демиелинизирующих нейропатий на основании наблюдавшейся демиелинизации нервных волокон в онтогенезе у мышей, нокаутных по этому гену.
Анализ сцепления заболевания с полиморфными маркерами из области локализации генов-кандидатов в семьях предпочтительнее, чем скрининг известных генов на наличие мутаций у отдельных пациентов, поскольку позволяет избежать ошибок, связанных с методами регистрации мутаций.
Таким образом, различные варианты поиска сцепления могут быть успешно применены в каждой конкретной ситуации.
- Читать далее "Строение хромосом человека. Классификация хромосом человека."
Оглавление темы "Анализ хромосом человека. Аномалии хромосом человека.":1. Картирование наследственных заболеваний. Методы картирования наследственных заболеваний.
2. Локусы генов. Генетическое расстояние между двумя локусами.
3. Строение хромосом человека. Классификация хромосом человека.
4. Анализ кариотипа человека. Цитогенетический метод анализа хромосом человека.
5. Молекулярно-цитогенетические методы анализа кариотипа человека. Нормальный кариотип человека.
6. Биохимические методы диагностики наследственных болезней. Иммуно-гистохимический метод.
7. Молекулярно-генетические методы диагностики наследственных заболеваний.
8. Хромосомные синдромы. Классификация хромосомных аномалий у человека.
9. Межхромосомные перестройки. Реципрокные транслокации. Робертсоновские транслокации.
10. Численные аномалии хромосом. Структурные аномалии хромосом.