
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизм развития (патогенез) тромботической микроангиопатии почек
Термин «тромботическая микроангиопатия» объединяет заболевания, проявляющиеся клинически микроангиопатической гемолитической анемией, тромбоцитопенией и во многих случаях почечной недостаточностью, а морфологически — тромбозом капилляров и артериол разных органов, в т.ч. почек. Важный диагностический признак — наличие в мазках периферической крови шистоцитов.
В отличие от ДВС тромботическая микроангиопатия обычно ассоциируется с нормальными показателями свертываемости крови и нормальными или немного повышенными уровнями продуктов деградации фибрина.
Классифицировать тромботическую микроангиопатию долгое время не удавалось из-за сходности клинических проявлений ее двух основных форм: ГУС и ТТП. В настоящее время известно, что заболевания ГУС и ТТП имеют несколько нозологических форм с разными этиологией, клиническим течением и терапевтическими подходами. Мы классифицируем ГУС и ТТП в соответствии с их этиологией и взаимосвязями по современным представлениям:
- типичный ГУС (синонимы: классический, эпидемический, диарея-позитивный) чаще всего обусловлен употреблением пищи, зараженной бактериями, продуцирующими шигаподобные токсины;
- атипичный ГУС (синонимы: неэпидемический, диарея-негативный), ассоциированный с:
- врожденными мутациями белков-регуляторов системы комплемента;
- повреждением эндотелия по различным причинам (наличие антифосфолипидных антител, осложнение беременности, сосудистые заболевания почек при склеродермии и гипертензии, применение оральных контрацептивов, химиотерапевтических, иммуносупрессивных препаратов и облучение);
- ТТП, которая ассоциируется с врожденным или приобретенным дефицитом ADAMTS 13 (металлопротеазы, регулирующей функцию WF).
а) Патогенез. При тромботической микроангиопатии превалируют два патогенетических механизма:
(1) повреждение и активация эндотелия (для ГУС);
(2) активация и агрегация тромбоцитов (для ТТП).
- Повреждение и активация эндотелия. При типичном ГУС триггером обычно является шигаподобный токсин, при врожденном атипичном ГУС — чрезмерная, неадекватная активация системы комплемента. Клиническая картина, напоминающая ГУС, может развиться в ответ на множество факторов, повреждающих преимущественно эндотелий. Повреждение эндотелия приводит к активации тромбоцитов и формированию тромбов в микроциркуляторном русле.
Есть доказательства, что снижение эндотелием синтеза простагландина I2 и оксида азота (оба являются ингибиторами агрегации тромбоцитов) сопровождается тромбозом. Снижение концентрации простагландина I2 и оксида азота и увеличение выработки эндотелина приводят к сужению сосудов, усугубляя гипоперфузию тканей. Кроме того, поврежденный эндотелий экспрессирует молекулы адгезии, рекрутирующие лейкоциты, которые также могут участвовать в развитии тромбоза.
- Активация и агрегация тромбоцитов. При ТТП происходят активация и агрегация тромбоцитов, вызванные крупными мультимерами WF, формирующимися из-за дефицита ADAMTS 13, в норме расщепляющей молекулы WF до меньшего размера. Дефицит ADAMTS 13 чаще всего обусловлен аутоантителами, ингибирующими ее функцию. Иногда хроническая рецидивирующая ТТП ассоциируется с врожденным дефицитом ADAMTS 13. Очень крупные мультимеры WF могут связываться с поверхностными гликопротеинами тромбоцитов и активировать их.
Этим можно объяснить спонтанное образование микротромбов в сосудистом русле. Независимо от пускового механизма при любой форме ГУС и ТТП тканевая дисфункция развивается, по-видимому, из-за микротромбов, обструкции сосудов и ишемии тканей.
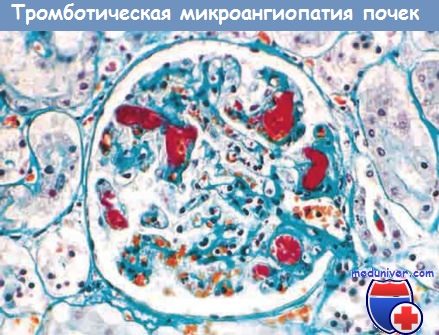
характерные для тромботических микроангиопатий (окрашивание на фибрин).
- Типичный гемолитико-уремический синдром. Это наиболее хорошо изученная форма ГУС. Большинство случаев заболевания обусловлено инфекцией ЖКТ бактериями Е. coli (чаще всего штаммом 0157:Н7). Эти бактерии вырабатывают токсин, напоминающий токсин Шига, выделяемый S. dysenteriae. Пути инфицирования различные, чаще всего употребление зараженных мясных продуктов (например, в гамбургерах), воды, цельного молока. Инфекция также может передаваться от человека к человеку контактным и бытовым путем. Однако большинство типичных ГУС, вызванных Е. coli, являются спорадическими. Заражение другими инфекционными агентами, включая S. dysenteriae, редко приводит к аналогичной клинической картине.
Типичный ГУС может наблюдаться у взрослых, особенно пожилых, но преимущественно развивается у детей. У последних ГУС — одна из самых частых при чин острой почечной недостаточности. После продромального периода с симптомами общего недомогания начинается острое кровотечение (кровавая рвота или мелена), развиваются выраженная олигурия, тромбоцитопения и гематурия, связанная с микроангиопатической гемолитической анемией. У некоторых детей присутствуют неврологические нарушения. В 50% случаев развивается гипертензия.
Шигаподобный токсин повреждает эндотелиальные клетки, индуцируя повышение экспрессии молекул адгезии лейкоцитов, повышение синтеза эндотелина, снижение образования оксида азота и апоптоз в присутствии таких цитокинов, как TNF. Эти изменения приводят к активации тромбоцитов, вазоконстрикции, вызывая характерную микроангиопатию. Есть доказательства того, что шигаподобный токсин также связывается с тромбоцитами и активирует их.
При типичном ГУС адекватное лечение почечной недостаточности с помощью диализа позволяет полностью у большинства пациентов восстановить функцию почек за несколько недель. Однако в случае тяжелых повреждений почек отдаленный прогноз (15-25 лет) менее благоприятен. В одном исследовании только у 10 из 25 пациентов, перенесших типичный ГУС, восстановилась функция почек, у 7 развилась хроническая почечная недостаточность.
- Атипичный гемолитико-уремический синдром. Такой ГУС наблюдается преимущественно у взрослых и имеет большое количество различных клинических вариантов. Более 50% таких пациентов имеют врожденный дефицит белков-регуляторов системы комплемента, чаще всего — фактора Н, в норме блокирующего C3bBb и защищающего клетки от повреждения при неконтролируемой активации системы комплемента. Небольшое количество пациентов имеют мутации двух других белков-регуляторов системы комплемента — фактора I и CD46. У пациентов с генетическими мутациями белков-регуляторов системы комплемента атипичный ГУС может развиться в любом возрасте.
У 50% пациентов он часто рецидивирует и прогрессирует до терминальной стадии хронической болезни почек. Поскольку дефицит белков-регуляторов системы комплемента присутствует у человека в течение всей его жизни, остается загадкой, почему атипичный ГУС манифестирует в разном возрасте. Вероятно, существуют неизвестные пока кофакторы, являющиеся триггерами развития ГУС.
С атипичным ГУС ассоциируются:
- антифосфолипидный синдром, первичный или вторичный при СКВ. В этом случае микроангиопатия имеет тенденцию к хроническому течению;
- беременность и послеродовой период. Послеродовая почечная недостаточность — вариант ГУС, развивающийся обычно в течение нескольких месяцев после разрешения неосложненной беременности. Состояние имеет неблагоприятный прогноз, лишь в легких случаях возможно полное восстановление функции почек;
- сосудистые заболевания, повреждающие почки (например, системный склероз и злокачественная гипертензия);
- химиотерапия или прием иммуносупрессивных препаратов (например, митомицина, циклоспоринов, цисплатина, гемцитабина);
- облучение почки.
Течение атипичного ГУС тяжелее, чем типичного, т.к. в большинстве наблюдений заболевание является вторичным, развивающимся на фоне хронической патологии, часто трудно поддающейся лечению. При атипичном ГУС, как и при типичном, у некоторых пациентов отмечается неврологическая симптоматика, но в отличие от ТТП уровень ADAMTS 13 в норме.
- Тромботическая тромбоцитопеническая пурпура. ТТП манифестирует классической пентадой: лихорадкой, нефрологическими симптомами, микроангиопатической гемолитической анемией, тромбоцитопенией и почечной недостаточностью. Как правило, ТТП вызвана антителами (аутоиммунными или индуцированными лекарственными препаратами) либо генетическими нарушениями, приводящими к функциональному дефициту ADAMTS13. Самая частая причина дефицита ADAMTS 13 — наличие ингибиторных аутоантител (чаще обнаруживают у женщин). Большинство пациентов молодые взрослые (до 40 лет).
Вовлечение нервной системы при ТТП — преобладающий клинический признак, в то время как почечную патологию наблюдают только у 50% больных. Клиническая картина характеризуется наличием микротромбов в артериолах всего организма. При отсутствии лечения заболевание имеет высокую летальность, однако иммуносупрессивная терапия и обменная трансфузия с антителами позволяют снизить летальность почти на 50%. Как и при ГУС, связанном с врожденным дефицитом белков-регуляторов системы комплемента, непонятно, почему при врожденном дефиците ADAMTS 13 заболевание проявляется только во взрослом возрасте. ТТП протекает, как правило, с периодами ремиссий и обострений.
б) Морфология. Морфологические изменения при разных формах ГУС и ТТП во многом похожи и больше зависят от длительности процесса, чем от этиологии.
В активной острой стадии заболевания при макроскопическом исследовании выявляют очаговый или диффузный кортикальный некроз и субкапсулярные петехии. При микроскопическом исследовании капилляры клубочка обтурированы тромбами из агрегатов тромбоцитов и небольшого количества фибрина. Стенки капилляров клубочка утолщены вследствие отека эндотелия и субэндотелиальных отложений клеточного детрита и фибрина. Результатом мезангиолизиса является разрушение матрикса и повреждение мезангиальных клеток. В междольковых артериях и артериолах часто возникают очаги фибриноидного некроза стенки и окклюзирующие тромбы.
Хроническое течение характерно для атипичного ГУС и ТТП, а морфологическая картина обусловлена длительным повреждением и попытками лечения заболевания. На коре почек обнаруживают рубцы различной давности. При световой микроскопии клубочки умеренно многоклеточны, а стенки капилляров значительно утолщены из-за расщепления базальной мембраны. В стенках артерий и артериол часто отмечают увеличение количества слоев клеток и соединительной ткани («луковичный» склероз), что приводит к сужению просвета сосудов, стойкой гипоперфузии и ишемической атрофии паренхимы, клинически проявляющимся почечной недостаточностью и гипертензией.
- Рекомендуем ознакомиться со следующей статьей "Механизм развития (патогенез) атеросклероза почек"
Оглавление темы "Заболевания почек":- Механизм развития (патогенез) доброкачественного нефросклероза
- Механизм развития (патогенез) злокачественного нефросклероза
- Механизм развития (патогенез) стеноза почечной артерии
- Механизм развития (патогенез) тромботической микроангиопатии почек
- Механизм развития (патогенез) атеросклероза почек
- Механизм развития (патогенез) серповидно-клеточной нефропатии
- Механизм развития (патогенез) диффузного кортикального некроза
- Механизм развития (патогенез) инфаркта почки
- Варианты врожденных аномалий почек
- Классификация кистозной болезни почек