
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Спастическая параплегия 1-го типа (SPG1) - клиника, диагностика
Х-сцепленные рецессивные наследственные спастические параплегии (НСП) относительно редки, но с точки зрения молекулярной генетики изучены раньше и полнее, чем аутосомно-доминантные и аутосомно-рецессивные формы параплегии.
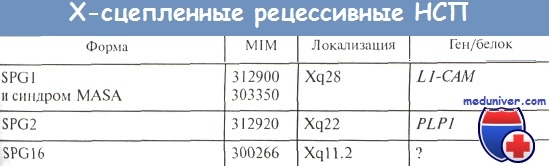
Спастическая параплегия 1-го типа (SPG1, MIM 312900) относят к осложненным вариантам НСП, хотя сопутствующие параплегии симптомы есть не во всех случаях. Болезнь характеризуется ранним началом и медленным прогрессированием симптомов спастичности с нормальной продолжительностью жизни. Основной из дополнительных (но не обязательных!) симптомов — умственная отсталость.
Возможны атаксия, атрофия зрительных нервов, а также характерное приведение больших пальцев рук из-за отсутствия (недоразвития) короткого или длинного разгибателей большого пальца. Например, в семье, где было проведено картирование гена болезни, у всех 6 больных имелась умственная отсталость (в том числе у 4 - тяжелая), а у 4 пациентов отсутствовал длинный разгибатель большого пальца (Kenwrick S. et al.).
Ген SPG1 картирован S. Kenwrick и соавт. на длинном плече Х-хромосомы в участке Xq28. В последующем сцепление с этим же локусом было обнаружено в семьях с синдромом MASA, а также в семьях с гидроцефалией, обусловленной стенозом сильвиева водопровода; на основании полученных данных было высказано предположение об аллельности этих заболеваний.
И действительно, когда соответствующий ген L1-САМ был идентифицирован (Jouet М. et al.), это предположение подтвердилось.
Продуктом гена L1-САМ (Cell Adhesion Molecule) является сложный мембраносвязанный гликопротеин, который широко представлен в нервной системе — в популяциях развивающихся и дифференцирующихся нейронов, а также в шванновских клетках. Белок L1-CAM является представителем суперсемейства молекул адгезии класса иммуноглобулина G и ответственен за созревание, миграцию нейронов и формирование их аксональных контактов с другими нейронами, клетками глии и мышцами в процессе онтогенетического развития либо после травмы (Sonderegger P., Rathjen Е., Jouet М. et al., Kenwrick S. et al.).
Исследованиями in vitro показано его участие в ветвлении аксонов, росте нервных окончаний и некоторых других процессах (Reid Е.). Такое многообразие функций свидетельствует о важной роли L 1-САМ в нейрогенезе и предполагает возможность множественных сочетанных аномалий развития при повреждении данного гена (что и подтверждается клинически — см. далее).
У мышей с «нокаутированным» геном L1-САМ закономерно развиваются спастичность и некоторые другие ассоциированные аномалии, включая анатомическую аномалию пирамидных трактов. В частности, у них наблюдается нарушение механизмов прокладывания корректного «пути» для растущих кортикоспинальных аксонов, что сопровождается отсутствием их перекреста в продолговатом мозге (Dahme М. et al., Fransen Е. et al.).
Такая уникальная патоморфология спастичности у L1-CAM-дефектных животных подчеркивает роль данного белка в обеспечении направленности аксонов кортикального двигательного нейрона.
Описано более 90 мутаций в гене L1-САМ, и почти каждая из них встретилась только в одной семье. Мутации распределяются довольно равномерно по всем 28 экзонам гена, более трети составляют миссенс-мутации, нередки также микроделеции и сплайсинговые мутации (Ruiz J. et al., De Angelis E. et al.).
Многие мутации подавляют экспрессию L1-CAM (т.е. действуют по механизму «потери функции»), некоторые миссенс-мутации влияют на те или иные функции белка (Kenwrick S. et al.). Имеются данные о зависимости фенотипа от вида мутации (Weller S. et al.).
Один из аллельных вариантов, связанных с мутациями гена L1-CAM - синдром MASA (Mental retardation, aphasia, Shuffling gait, Aiducted thumbs; синдром умственной отсталости, афазии, «шаркающей» походки и приведенных больших пальцев рук). Синдром MASA (другое название — синдром Таре—Мейсона) может быть отнесен к осложненным формам НСП, поскольку составной частью синдрома является спастический парапарез («шаркающая» походка).
Отсутствие или недоразвитие разгибателей большого пальца, встречающиеся при SPG1, для синдрома MASA особенно типичны. Умственная отсталость обычно легкая или умеренная; кроме основных симптомов возможны гидроцефалия и низкорослость.
Известны еще два аллельных L1-CAM-ассоциированных фенотипа — гидроцефалия со стенозом сильвисва водопровода и гипоплазия мозолистого тела; при обоих синдромах также возможно развитие спастической параплегии.
Все четыре состояния, частично перекрывающиеся по своему фенотипу (иногда и у членов одной семьи), нередко объединяют в единый клинический континуум под названием синдрома CRASH (Callosal hypoplasia, mental Retardation, Alductcd thumbs, Spastic paraparesis, Hydrocephalus; гипоплазия мозолистого тела, умственная отсталость, спастический парапарез, гидроцефалия) (Fransen Е. et al.).
При SPG1 и разнообразных ее аллельных вариантах в принципе возможна прямая ДНК-диагностика, но на практике ее затрудняют многообразие мутаций и отсутствие мажорных мутаций в гене L1-CAM, а также распределение мутаций практически по всему протяженному гену.
- Читать "Спастическая параплегия 2-го типа (SPG2) - клиника, диагностика"
Оглавление темы "Наследственные спастические параплегии (НСП)":- Спастическая параплегия 1-го типа (SPG1) - клиника, диагностика
- Спастическая параплегия 2-го типа (SPG2) - клиника, диагностика
- Спастическая параплегия 16-го типа (SPG16) - клиника, диагностика
- Синдром Шегрена-Ларссона - клиника, диагностика
- Пример синдрома Шегрена-Ларссона
- Спастическая атаксия типа Шарлевуа-Сагенэ - клиника, диагностика
- Ранний восходящий спастический паралич и ювенильный первичный боковой склероз - клиника, диагностика
- Нарушение распознавания клеток и клеточных сигналов L1-CAM при НСП
- Нарушение процессов миелинизации при НСП: протеолипидный белок и его изоформы
- Нарушения в системе митохондриальных молекулярных шаперонов при НСП и HSP60