
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Пример атипичной формы атаксии Фридрейха
Семья Аг.-Сеид. (см. родословную). В данной уникальной туркменской инбредной семье имело место крайне редкое псевдодоминантное наследование атаксии Фридрейха. Родители больных сибсов — троюродные брат и сестра, причем отец болен атаксией Фридрейха, а мать, как показал ДНК-анализ (см. дорожку Ш-2 электрофореграммы), является гетерозиготным носителем мутации.
При этом у 2 наблюдавшихся нами сестер диагностирована атаксия Фридрейха с очень поздним началом и «мягким» прогрессированисм (фенотип VLOFA), тогда как у их родных братьев — «классический» ранний фенотип заболевания с быстрым и тяжелым течением.
Больная Аг.О., туркменка, 50 лет (IV-2). Считает себя больной с 44-летнего возраста, когда появилась прогрессирующая шаткость при ходьбе, постепенно нарастала слабость в ногах и затем в руках, присоединились замедленность и нечеткость речи, а в последние 3 года — боли в области сердца. При осмотре отмечены «ладьевидные» изменения фаланг пальцев обеих рук, «полая стопа», кифосколиоз.
В неврологическом статусе: умеренно выраженная дизартрия; непостоянный статический тремор головы и дистальных отделов правой руки; небольшое снижение силы в мышцах конечностей (преимущественно в кистях и стопах); ограничение объема движений в голеностопных суставах; гипотрофия межостных мышц кистей и m. hypothenar; диффузная мышечная гипотония; отсутствие сухожильных рефлексов; симптом Бабинского с двух сторон.
Гиперметрия в руках, замедление темпа движений в пробе на диадохокинез; интенционный тремор в конечностях при выполнении координаторных проб; легкая атаксия туловища в положении сидя с закрытыми глазами; неустойчивость в пробе Ромберга; больная способна самостоятельно ходить в пределах помещения, при ходьбе пошатывается в стороны. Умеренная гипестезия по полиневритическому типу в ногах, негрубое нарушение суставно-мышечного чувства в пальцах стоп, вибрационная чувствительность в руках резко снижена (до 5 с), в ногах отсутствует.
При дополнительном лабораторно-инструментальном обследовали выявлены следующие изменения: феномен лактат-ацидоза в крови; нарушение активности ферментов-дегидрогеназ; на ЭКГ — вменения миокарда боковой стенки левого желудочка (признаки миокардиодистрофии по заключению терапевта); на компьютерных томограммах головного мозга — небольшие атрофические изменения ствола мозга, верхнего червя мозжечка и больших полушарий; признаки генерализованной демиелинизирующей сенсорной невропатии по данным электронейромиографии.
Результаты ДНК-диагностики: выявлено носительство 2 мутантных аллелей гена FRDA с числом GAA-повторов 250 и 700 копий (см. электрофореграмму, дорожка IV-2). Диагноз: атаксия Фридрейха, атипичная форма с очень поздним началом (вариант VLOFA).
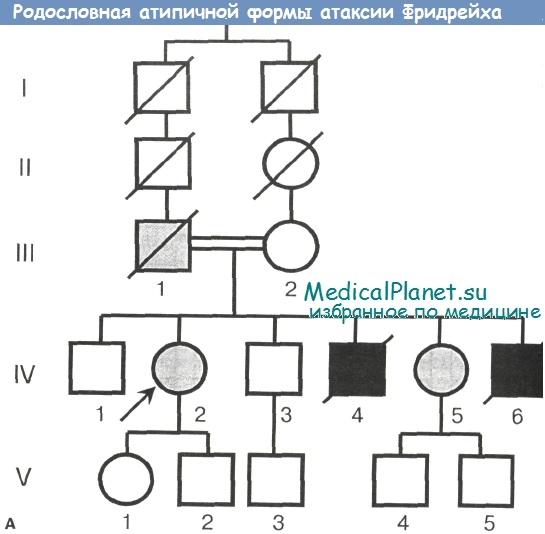
а - родословная семьи, демонстрирующая псевдодоминантное наследование болезни в двух поколениях (черным цветом обозначены больные с тяжелым «классическим» фенотипом болезни, серым цветом — больные с «мягким» поздним фенотипом атаксии Фридрейха)
Родная сестра пробанда — больная Сеид. Г., 57 лет (IV-5), с 48-летнего возраста отмечает нарастающую шаткость при ходьбе, к которой спустя несколько лет постепенно присоединилась слабость в ногах, а позднее и в руках, похудание мышц конечностей, появились изменения речи. В последние годы отмечается дрожание в руках, боли в области сердца, одышка при ходьбе. При осмотре: кифосколиоз позвоночника в грудном отделе, умеренная деформация кистей (по типу «полой кисти») и стоп (стопы Фридрейха).
Неврологический статус: мелкоамплитудный горизонтальный нистагм; дизартрия, дисфония. Легкое снижение силы в мышцах рук (больше в кистях), отчетливо выраженный парез проксимальных и дистальных отделов ног с ограничением объема движений в голеностопных суставах; гипотрофия мелких мышц кистей рук, диффузная гипотрофия мускулатуры ног; мышечная гипотония; сухожильная арефлексия; симптом Бабинского с двух сторон.
Грубая гиперметрия и адиадохокинез в руках; координаторные пробы в верхних и нижних конечностях выполняет с выраженной дискоординацией и интенционным тремором; грубая туловищная атаксия в положении сидя и стоя в пробе Ромберга с открытыми и закрытыми глазами; стоять и ходить в пределах помещения может только с поддержкой из-за атаксии и пареза в ногах. Умеренная гипестезия в дистальных отделах ног; нарушение суставно-мышечного чувства в пальцах стоп, вибрационная чувствительность в руках резко снижена (до 5 с), в ногах отсутствует.
Данные дополнительных лабораторно-инструментальных методов обследования: феномен лактат-ацидоза в крови; нарушение активности ферментов-дегидрогеназ; на ЭКГ - метаболические изменения миокарда нижней стенки левого желудочка (признаки миокардиодистрофии согласно заключению терапевта), признаки генерализованной демиелинизирующей сенсорной невропатии по данным электронейромиографии.
Результаты ДНК-диагностики: выявлено носительство 2 мутантных аллелей гена FRDA с числом GAA-повторов 250 и 700 копий (см. электрофореграмму, дорожка IV-5).
Диагноз: атаксия Фридрейха, атипичная форма с очень поздним началом (вариант VLOFA).
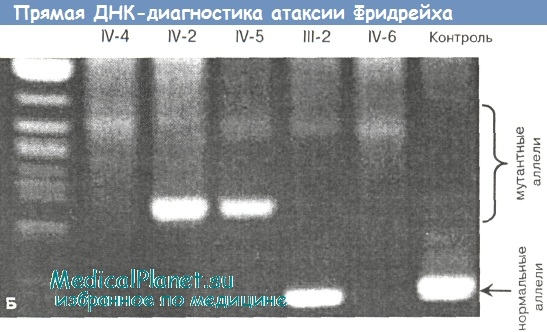
б - результаты прямой ДНК-диагностики.
В описанных случаях обращает на себя внимание весьма поздний дебют заболевания — в возрасте 45 и 48 лет, а также относительно медленное течение болезни (так, больная Аг.О. на протяжении свыше 8 лет с момента дебюта заболевания все еще способна самостоятельно передвигаться). Аналогичная клиника, согласно данным анамнеза, имела место у отца больных, заболевшего в 26-летнем возрасте, утратившего способность ходить к 45 годам, прикованного к креслу к 60 годам и умершего в возрасте 64 лет.
В то же время, еще у двоих больных братьев в данной семье (IV-4 и IV-6), в противоположность характеру болезни у сестер и отца, заболевание дебютировало в возрасте 8 и 10 лет, причем клиническая картина у них характеризовалась развитием тяжелого «классического» фенотипа атаксии Фридрейха и относительно ранней смертью.
Причины такого выраженного внутрисемейного полиморфизма болезни в данном наблюдении стали ясны после проведения молекулярно-генетического анализа. Оказалось, что обе сестры являются носителями двух различных аллелей гена FRDA - с тяжелой и легкой степенью экспансии GAA-повторов (соответственно 700 и 250 копий). В данной комбинации более длинный аллель унаследован от матери, а более короткий (т.е. относительно сохранный и определяющий развитие более «благоприятного» позднего фенотипа) — от отца.
Очевидно, что аналогичная комбинация аллелей с тем же «благоприятным» фенотипом имела место и у отца больных, причем длинный аллель у него и у матери является генетически общим и унаследован от одного предка. Напротив, больные братья, как показывает результат ДНК-анализа, унаследовали от отца «неблагоприятный» аллель FRDA (700 копий GAA-триплеов), что в комбинации с таким же длинным материнским аллеем привело к гомозиготности по тяжелой экспансии GAA-повторов и манифестации «классического» раннего фенотипа атаксии Фридрейха.
Можно с уверенностью утверждать, что без применения методов ДНК-анализа адекватная диагностика болезни в данной уникальной семье была бы практически невозможной.
- Читать "Пример атипичной формы атаксии Фридрейха варианта FARR"
Оглавление темы "Атаксия Фридрейха":- Пример атипичной формы атаксии Фридрейха
- Пример атипичной формы атаксии Фридрейха варианта FARR
- Пример атаксии Фридрейха фенотипа спастической атаксии
- Пример мягкой формы атаксии Фридрейха - акадской формы
- Морфология атаксии Фридрейха
- Дополнительные методы обследования при атаксии Фридрейха
- Дифференциальная диагностика атаксии Фридрейха
- ДНК-диагностика атаксии Фридрейха
- Принципы лечения атаксии Фридрейха и ее эффективность
- Профилактика и скрининг на атаксию Фридрейха