
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Метаболические энцефалопатии детей-подростков: болезнь Вильсона-Коновалова, хорея Гентингтона, синдром Ханта
В эту группу включены редкие наследственные и семейные заболевания нервной системы с поздней манифестацией (4-15 лет и более) и медленным, но прогрессирующим течением. Этиология и патогенез многих из них до сих пор остаются неясными и диагностика обычно базируется на сугубо клинических симптомах.
Обнаруживаемые биохимические изменения часто носят неспецифический характер. Для большинства заболеваний характерно преимущественное поражение экстрапирамидной системы или же координаторных и пирамидных систем мозга. С целью облегчения дифференциально-диагностического процесса мы разделили эти заболевания на небольшие подгруппы: семейные болезни с паркинсонизмом, семейные болезни с дистонией или хореоатетозом, редкие семейные экстрапирамидные болезни, семейные метаболические энцефалопатии с диффузным поражением мозга, прогрессирующие мозжечковые атаксии, полиомиоклонии, семейные полиневропатии.
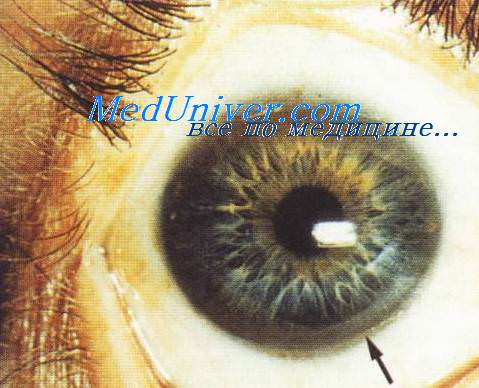
1. Семейная гепатоцеребральная дегенерация (болезнь Вильсона-Коновалова, псевдосклероз Штрюмпеля-Вестфаля). Различают 4 формы заболевания: абдоминальную (преимущественно нарушение функции печени и желудочно-кишечного тракта), ригидно-аритмо-гиперкинетическую (преобладают гиперкинезы и изменения мышечного тонуса), дрожательно-ригидную и экстрапирамидную.
Манифестация неврологических симптомов наступает не раньше, чем в 7-10 лет: нарастает мышечная ригидность, появляются торсионно-дистонические или атетоидные гиперкинезы, дизартрия, дисфагия, снижение интеллекта, псевдо-бульбарные расстройства. Изменения печени расцениваются как подострый гепатит (анорексия, диспепсия, рвота, гипертермия, ремиттирующая желтуха, затем цирроз печени). При патоморфологических исследованиях обнаруживаются отложения меди в головном мозге, печени, селезенке, почках и др.
В мозге выявляются очаги сморщивания и склерозирования мозговой ткани, разрастание глии и образование кист в коре и подкорковых образованиях. В основе заболевания - нарушение синтеза белка церулоплазмина. Тип наследования — А/Р. Дифференциальный диагноз проводится с другими наследственными энцефа-лопатиями с экстрапирамидной симптоматикой, с другими формами цирроза печени.
2. Хорея Гентингтона (детская форма). Манифестация возможна в возрасте 3 лет, но чаще между 5 и 12 годами: нарастает интеллектуальный дефект, нарушается речь, появляются блефароспазм, паркинсонизм, интенционный тремор, зрительные расстройства (оптокинетический нистагм), пирамидные знаки, хореоатетоз. Болезнь медленно прогрессирует (5-15 лет). При патоморфологическом исследовании обнаруживается атрофия мозга с преимущественным поражением лобных долей и подкорковых образований. Патогенез заболевания неясен, относится к дегенеративным болезням с аутосомно-доминантным типом наследования. Дифференциальный диагноз проводится со всеми прогрессирующими экстрапирамидными болезнями, психозами и болезнями, сопровождающимися деменцией.
3. Синдром Ханта (миоклонус-атаксия). Считается типичным паркинсонным синдромом. Манифестация заболевания возможна в 10 лет и более: тремор, миоклонии, брадикинезии, маскообразное лицо, мышечная гипотония, нистагм, атаксия. При патоморфологическом исследовании обнаруживают дегенерацию красного и зубчатого ядер мозжечка. Патогенез остается неясен. Тип наследования — А/Р. Дифференциальный диагноз проводится с миоклонус-эпилепсией и болезнью Фридрейха.
- Рекомендуем далее ознакомиться со статьей "Метаболические энцефалопатии детей-подростков: прогрессирующая торсионная дистония, болезнь Галлервордена-Шпатца, синдром хореоатетоза"
Оглавление темы "Метаболические энцефалопатии у детей":- Метаболические энцефалопатии детей ясельного возраста: метахроматическая лейкодистрофия
- Метаболические энцефалопатии детей ясельного возраста: болезнь Аустина, нейроаксональная дегенерация, синдром Луи Бара
- Метаболические энцефалопатии детей ясельного возраста: болезни Нимана-Пика, Дери, Краббе, Гоше
- Метаболические энцефалопатии детей ясельного возраста: нейрональный цероидлипофусциноз, синдром Альперса
- Дифференциация метаболических энцефалопатий детей ясельного возраста
- Метаболические энцефалопатии детей-подростков: болезнь Вильсона-Коновалова, хорея Гентингтона, синдром Ханта
- Метаболические энцефалопатии детей-подростков: прогрессирующая торсионная дистония, болезнь Галлервордена-Шпатца, синдром хореоатетоза
- Метаболические энцефалопатии детей-подростков: атрофия Corpus Luisi, лейкодистрофии
- Метаболические энцефалопатии детей-подростков: атаксия Фридрейха, синдромы Русси-Леви, Ван-Богарта-Шредера-Эпштейна
- Метаболические энцефалопатии детей-подростков: синдром Базена-Корнцвейга, миоклоническая эпилепсия типа Лафора