
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Кожа при амилоидозе - клиника, диагностика
а) Анамнез. У большинства пациентов с системным амилоидозом клиническая картина является неспецифичной, и поэтому амилоидоз часто становится неожиданной находкой при биопсии пораженного органа.
б) AL-амилоидоз. AL-амилоидоз обычно встречается у пациентов в возрасте старше 50 лет, хотя может развиваться и у взрослых очень молодого возраста. В большинстве случаев лежащий в основе плазменный клон клеток не обнаруживается до тех пор, пока у пациента не разовьется связанное с амилоидозом нарушение функции органа. Клинические проявления чрезвычайно разнообразны, так как почти любой орган, за исключением головного мозга, может быть непосредственно вовлечен в патологический процесс.
Хотя определенные клинические признаки, такие как макроглоссия и окологлазничные экхимозы, с наибольшей вероятностью указывают на AL-амилоидоз, и часто встречается нарушение функций множественных органов, у многих пациентов отмечаются неспецифические симптомы, такие как недомогание и потеря веса.
в) Системный АА-амилоидоз. АА-амилоидоз может возникать в любом возрасте как у детей, так и у взрослых, при этом средний возраст развития заболевания в Великобритании составляет 48 лет. Почти 95% пациентов ранее ставился диагноз хронического воспалительного заболевания, такого как ревматоидный артрит или любого другого воспалительного поражения кожи ревматического происхождения; хронический сепсис, обычно при бронхоэктазах, осложнения параплегических состояний, или употребление наркотиков.
Несколько чаще встречается у мужчин. Несмотря на то, что заболевание может развиться довольно остро, средний латентный период между появлением первых признаков хронического воспалительного заболевания и клинически значимого амилоидоза составляет почти 20 лет. Амилоидоз обычно имеет симптомы протеинурии с последующей прогрессирующей почечной недостаточностью, часто в сочетании с нефротическим синдромом.
г) Наследственный амилоидоз. Клиническая картина наследственного амилоидоза различается, в зависимости от варианта белка и мутации, и даже среди кровных родственников. Тяжелая прогрессирующая периферическая и/или вегетативная нейропатия является главным признаком наследственного TTR-амилоидоза (семейная амилоидная полинейропатия), но также распространено поражение сердца. Аполилипопротеин-А1 -амилоидоз иногда становится причиной нейропатии, но это не является характерной чертой других наследственных типов амилоидоза, при которых обычно поражаются внутренние органы. Даже при доминантном наследовании мутации белка-предшественника амилоида (амилоидогена) заболевание проявляется не в каждом поколении из-за различной пенетрантности гена.

д) Кожные проявления амилоидоза:
1. Кожа при системном AL-амилоидозе. Кожные проявления типичны для системного амилоидоза, особенно AL-типа, и, как сообщается, встречаются у 40% пациентов. Поражения обычно отражают капиллярную инфильтрацию и ломкость и проявляются петехиями и пурпурой, особенно поражая веки, область роста бороды и верхнюю часть груди. Ксантоматозные папулы или бляшки также наблюдаются довольно часто, получены данные о редких амилоидозных гиперпигментированных кератозных элементах. Термин «щипковая пурпура» описывает феномен, при котором если амилоидную бляшку ущипнуть или потереть, возникает кровоизлияние в ткань. К другим редким кожным признакам относятся склеродермоподобные изменения, алопеция и дистрофия ногтей. У нескольких пациентов был описан буллезный амилоидоз.
Буллы на коже и слизистых оболочках могут быть интрадермальными или субэпидермальными и представляют собой напряженные, часто геморрагические волдыри. Генерализованная инфильтрация кожных тканей часто вызывает утолщение кожи с утратой лицевых морщин и может ограничивать открытие рта.
2. Кожа при наследственном амилоидозе. Лисозим-амилоидоз часто вызывает петехиальную сыпь, а апо-липопротеин-А1-амилоидоз может проявляться в виде желтоватых инфильтрированных бляшек и поражений по типу акантозиса нигриканс. Кожные симптомы при семейной амилоидной полинейропатии, по-видимому, сильно связаны с периферической нейропатией и у 142 португальских пациентов включали в себя в 82% случаев ксероз, в 22% — себорейный дерматит, в 20% — травматические и ожоговые поражения, в 14%—нейропатические язвы и в 10,5% — онихомикоз.
3. Кожа при локализованном кожном AL-амилоидозе. Как и при системном AL-амилоидозе, фибриллы при локализованном AL-амилоидозе происходят из N-терминаль-ных расщепленных фрагментов легких цепей моноклонального иммуноглобулина. Поражения такие же, как и при системном AL-амилоидозе, и представлены единичными или, наиболее часто, множественными элементами, расположенными на любом участке кожи. Среди наблюдавшихся нами 20 пациентов с таким диагнозом у 35% обнаружена медленно растущая узелковая форма, узловая или ксантоматозная, у 20% — пурпура, еще у 20% — кожный зуд, у 15% — местные кровотечения, у 10%— локализованный отек.
4. Кожа при макулярном амилоидозе и лишайном амилоидозе. Макулярный и лишайный амилоидоз представляют собой варианты одного патологического состояния, при котором волокна амилоида образуются из кератина после поражения эпидермиса и апоптоза кератиноцитов. Этот процесс обычно идиопатический или вызван трением, но есть данные и о связи этого заболевания с болезнями соединительной ткани (первичным билиарным циррозом и системной красной волчанкой), а также (у нескольких кровных родственников) с врожденной пахионихией и множественными эндокринными неоплазиями 2а типа.
Макулярный амилоидоз обычно поражает верхнюю часть спины и конечности и может персистировать в течение многих лет. Сыпь сопровождается кожным зудом в большинстве случаев и представляет собой небольшие коричневые пятна, распространенные как рябь на поверхности воды. Среди больных могут преобладать женщины, заболевание больше распространено среди жителей Центральной и Южной Америки, Среднего Востока и азиатских стран, за исключением Китая. Отложения амилоида обычно граничат с сосочковым слоем дермы и не поражают кровеносные сосуды и придаточные структуры. Ранние элементы содержат небольшие многогранные аморфные глобулы в пределах сосочков, и без применения специальных методов окрашивания такие поражения легко можно не заметить.
Поздние поражения представляют собой глобулы, которые сливаются, распространяясь на сосочек, смещая латерально нервные сплетения. Лишайный амилоидоз—это самый распространенный тип кожного амилоидоза среди китайцев и обычно поражающий взрослых. Клинически характеризуется множественными интенсивно зудящими несливающимися гиперкератическими папулами на разгибательных поверхностях нижних конечностей, которые при шелушении образуют серо-коричневые чешуйки. Возможно распространение на разгибательные поверхности рук и туловище. Гистологически отложения амилоида очень сходны с таковыми при макулярном амилоидозе, но имеют незначительно более крупный размер и сопровождаются акантозом неправильной формы и гиперкератозом надлежащего эпидермиса.
5. Другие типы локализованного кожного амилоидоза. Хорошо изучены отложения незначительных микроскопических количеств амилоида в сочетании с разнообразными кожными поражениями. К документированным предрасполагающим состояниям относятся интрадермальные невусы, опухоли потовых желез, пиломатриксома, дерматофиброма, себорейный кератоз, солнечный эластоз, фоточувствительная аннулярная эластолитическая гигантоклеточная гранулема, актинический кератоз, порокератоз Мибелли, амилоидоз кожи, болезнь Боуэна, базально-клеточный рак, осложнения PUVA-терапии.
6. Аносакральный кожный амилоидоз. Это редкий синдром, описанный среди мужчин в Японии и Китае, при котором образуются пигментные пятна и лоснящиеся гиперкератические очаги, радиально расходящиеся из области ануса.
7. Амилоидоз, вызванный инсулином. Редкое осложнение длительной инсулинотерапии, возникающее в месте повторяющихся инъекций инсулина. Вероятна ошибочная диагностика опухоли.
8. Семейный первично локализованный амилоидоз кожи. Исключительно редкое аутосомно-доминантное заболевание, характеризующееся хроническим зудом, возникающим в детском или раннем подростковом возрасте, гиперкератическими папулами и/или гиперпигментацией на конечностях или туловище. Считается относительно распространенным заболеванием в Юго-Восточной Азии. Вероятнее всего поражает только кожу. Появление амилоидоза ассоциировано с мутацией в генах OSMR и IL31RA, относящихся к семейству интерлейкина-6 рецептора цитокина.
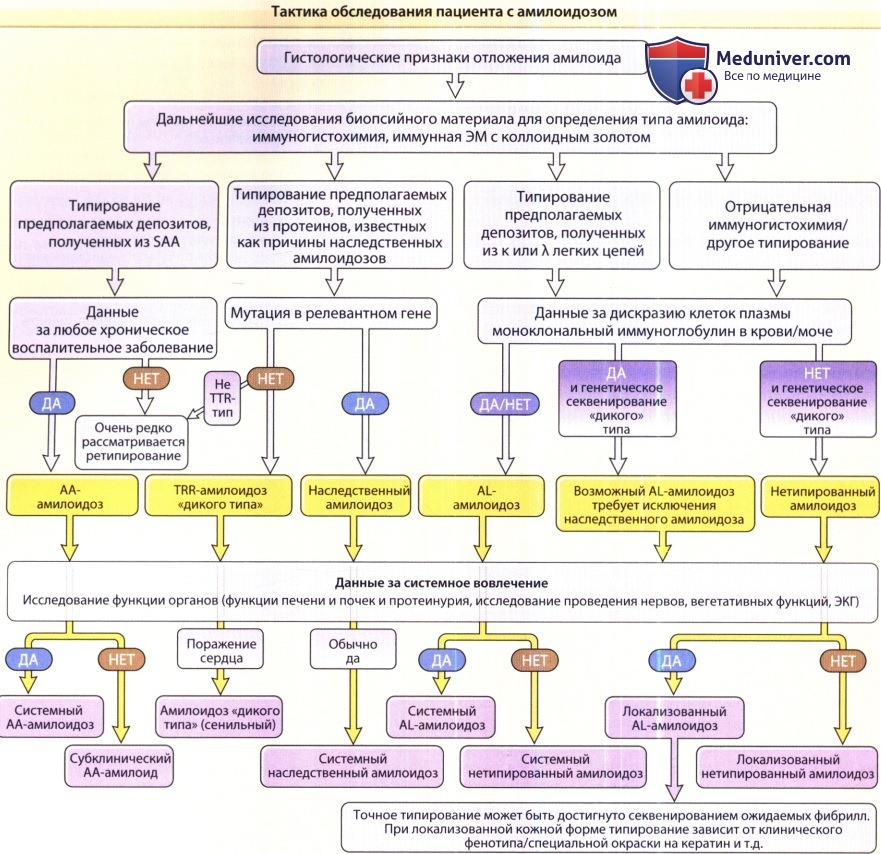
Ведение больного полностью зависит от точного типирования. Это является как высоко специализированной, так и комплексной задачей.
Точная установка типа амилоидоза предполагает ряд специальных тестов и лучше всего проводится в центрах, имеющих опыт таких исследований.
КМ — костный мозг; ЭКГ—электрокардиография; ЭМ — электронная микроскопия;
SAA—сывороточный протеин амилоид A; SAP — сывороточный компонент амилоид P; TTR — транстиретин.
е) Объективное обследование при амилоидозе:
1. Системный AL-амилоидоз. Системный AL-амилоидоз многообразен в своих проявлениях, и способен вызывать симптомы поражения любых органов и систем, исключая головной мозг.
2. Системный АА-амилоидоз. Этот тип почти у половины пациентов проявляется нефротическим синдромом. Часто встречается спленомегалия, гепатомегалия выявляется в 10% случаев. Редко отмечается поражение сердца или вегетативная нейропатия.
3. Наследственный амилоидоз. Физикальные данные при наследственном амилоидозе зависят от белкового варианта заболевания и специфических мутаций и могут различаться даже в пределах отдельных семей.
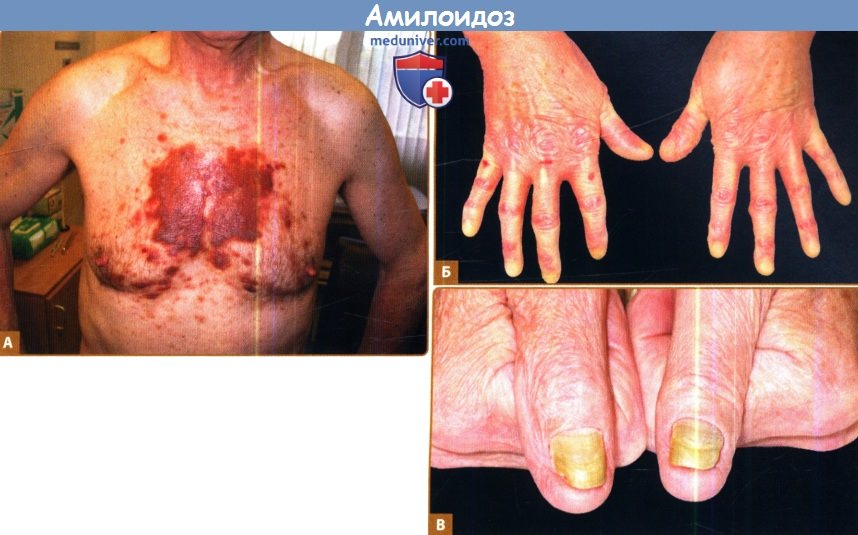
А. Пурпура, обусловленная ломкостью капилляров.
Б. Склеродермоподобные изменения на руках.
В. Ногтевая дистрофия.
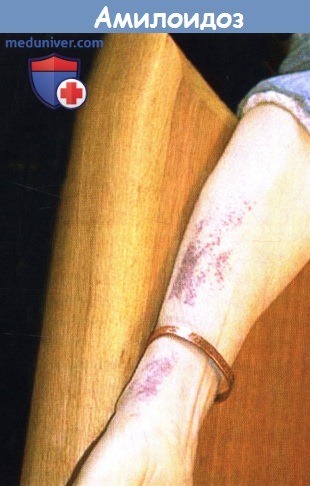
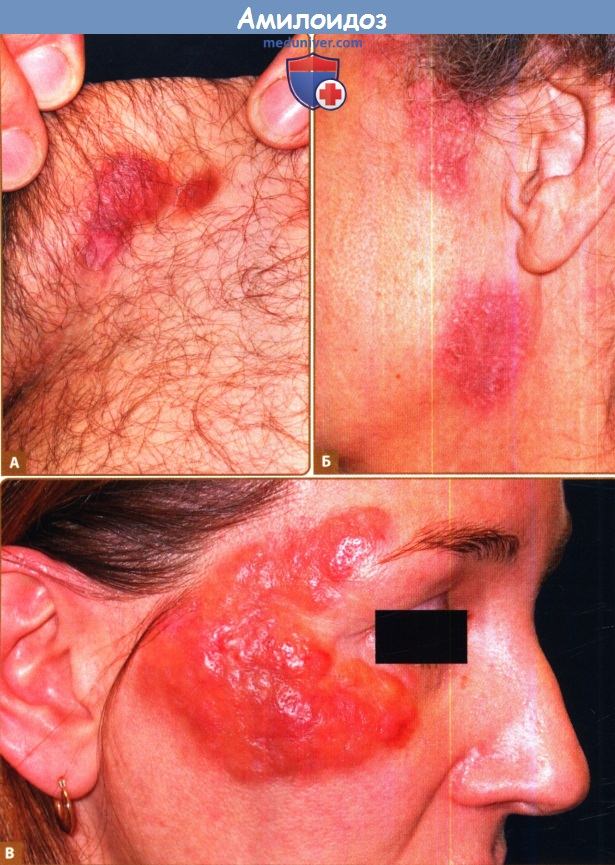
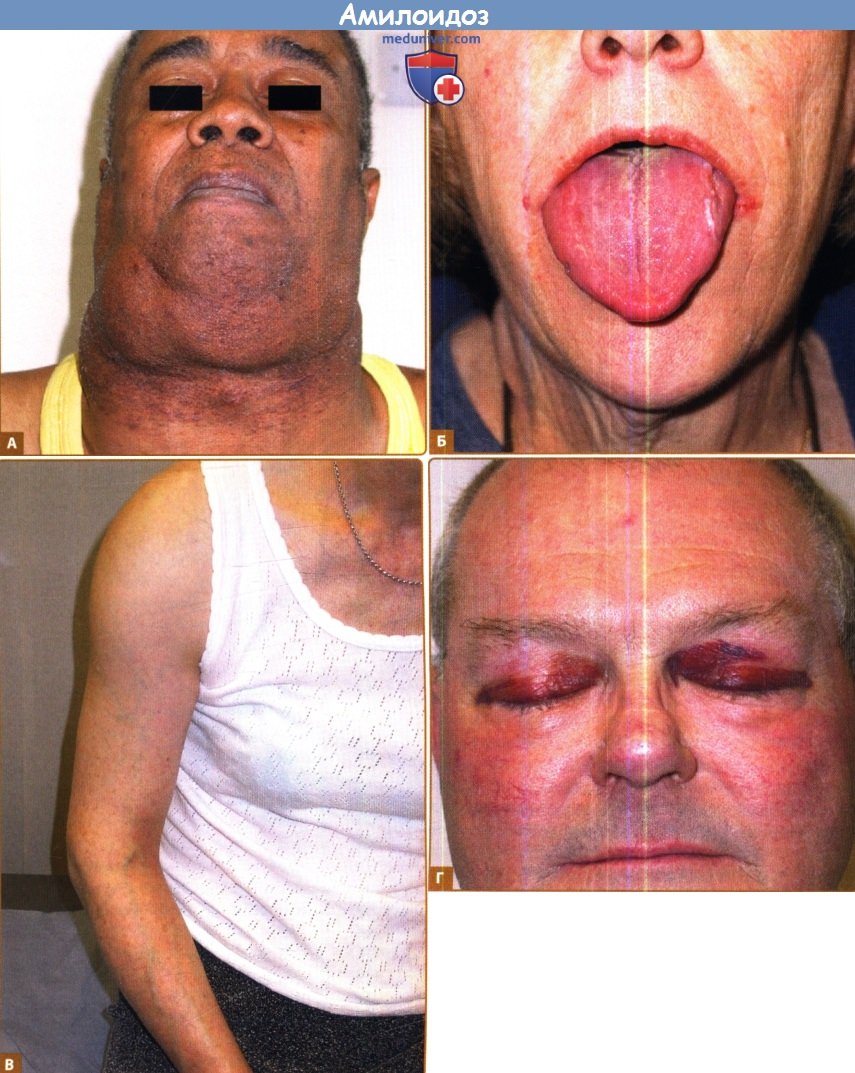
А. Лимфаденопатия. Б. Макроглоссия.
В. Мышечная псевдогипертрофия. Г. Периорбитальная пурпура.
ж) Анализы при амилоидозе. Во всех случаях необходима всесторонняя оценка почечных и печеночных функций, включая анализы крови и оценку клиренса креатинина и протеинурии. Необходимо оценить дискразию клеток плазмы путем электрофореза и иммунофиксации сыворотки и мочи. При подозрении на амилоидоз AL-типа обычно необходим забор образцов костного мозга. При возможности таких исследований для диагностики AL, SAA и АА-амилоидоза очень ценными являются анализы сыворотки на потенциальные фибриллярные белки-предшественники и свободные легкие цепи иммуноглобулинов.
з) Гистология амилоидоза. Выявление амилоида зависит от патогномоничного красно-зеленого дихроизма, наблюдаемого, когда ткань при окрашивании анилиновым красителем Конго красным становится видимой под кросс-поляризованным светом. Этот оптический эффект вызван рядом молекул красителя, расположенным вдоль фибрилл. Связывание тиофлавина Т обычно соотносится с двойным лучепреломлением Конго красного, но является менее специфичным. Окрашивание Конго красным на амилоид является не очень чувствительным тестом и требует наличия достаточного количества амилоида, толстых срезов ткани, правильного окрашивания и визуализирующих процедур, а также соответствующего опыта исследователя.
В негативно окрашенных микроскопических образцах волокна амилоида обычно достигают около 10 нм в диаметре, являются прямыми, твердыми, неветвящимися, средней длины и состоят из переплетенных протофибрилл.
Положительные результаты гистологического исследования на амилоид могут сопровождаться иммуногистохимическими тестами для оценки типа фибриллярного протеина. Необходимые антитела являются широко доступными, но хотя иммуногистохимический анализ обычно приносит четкие результаты в случае АА-амилоидоза, при отложениях AL-амилоида он часто бывает неэффективным. Экспертная оценка иммуногистохимического типирования амилоида при наследственном заболевании ограничена, и точное иммуногистохимическое типирование отложений амилоида достигается нечасто. Прямое секвенирование выделенных волокон позволяет проводить идентификацию типа амилоида, но требуемые для этого технологии обычно доступны лишь в исследовательских учреждениях.

А. Окрашивание Конго красным.
Б. Тот же срез под кросс-поляризованным светом с красно-зеленым двойным лучепреломлением.
и) Генетика амилоидоза. Поиск амилоидогенных мутаций должен проводиться у всех пациентов с неустановленным типом амилоида, так как наследственный амилоидоз составляет существенную часть этих случаев.
к) Методы определения амилоида in vivo:
1. SAP-сцинтиграфия (SAP — SERUM AMYLOID Р-COMPONENT — СЫВОРОТОЧНЫЙ Р-КОМПОНЕНТ АМИЛОИДА). SAP — это хорошо сохранный невариабельный гликопротеин плазмы семейства пентраксина, который становится специфическим и высоко концентрированным в отложениях амилоида всех типов в результате его кальций-зависимого связывания с волокнами амилоида. Для диагностики и количественного мониторинга отложений амилоида может использоваться радиоизотопная SAP-сцинтиграфия. Этот безопасный и неинвазивный метод предоставляет данные о наличии, распространении и объеме отложений амилоида во внутренних органах (за исключением сердца), и серийное сканирование позволяет контролировать прогрессирование заболевания и ответ на терапию. К сожалению, метод неэффективен для определения локализации отложений амилоида в сердце и малодоступен в связи с высокой стоимостью.
2. Исследование сердца. Амилоидоз сердца лучше всего оценивается при эхо- и электрокардиографии. Допплер-эхокардиография в двух проекциях выявляет концентрические бивентрикулярные утолщения стенок с рестриктивным типом наполнения. Амилоид вызывает диастолическую дисфункцию с сохранением сократимости вплоть до самой последней стадии. У пациентов с выраженным амилоидозом сердца электрокардиография может быть нормальной, но при прогрессировании заболевания обычно выявляются низкий вольтаж, патологические зубцы Q (псевдоинфарктный тип). Более новые технологии магнитно-резонансной томографии могут также способствовать оценке тяжести амилоидоза сердца, точно так же, как серологические исследования на сердечные биомаркеры N-терминального промозгового натрийуретического пептида и тропонина Т.

- Рекомендуем далее ознакомиться со статьей "Течение и прогноз амилоидоза"
Редактор: Искандер Милевски. Дата публикации: 2.11.2018