
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Симптомы и клиника порокератоза
а) Порокератоз Мибелли. В типичных случаях начало порокератоза Мибелли приходится на младенческий или детский возраст. Заболевание проявляется в виде бессимптомных кольцевидных папул коричневого или телесного цвета с характерным приподнятым краем. Четко очерченный гиперкератотический край, высота которого, как правило, превышает 1 мм, имеет характерную продольную бороздку.
Центральная часть очага может быть гиперпигментированой, гипопигментированной, вдавленной, атрофической или ангидротической. Диаметр очагов составляет от нескольких миллиметров до нескольких сантиметров, однако в редких случаях наблюдаются гигантские очаги диаметром до 20 см, локализованные преимущественно на голенях и стопах. У пациентов с крупными очагами повышен потенциал злокачественности.
Иногда развиваются множественные очаги, которые, как правило, имеют одностороннюю и ограниченную определенным участком локализацию. Заболевание наследуется по аутосомно-доминантному типу. Очаги персистируют неопределенно долго.
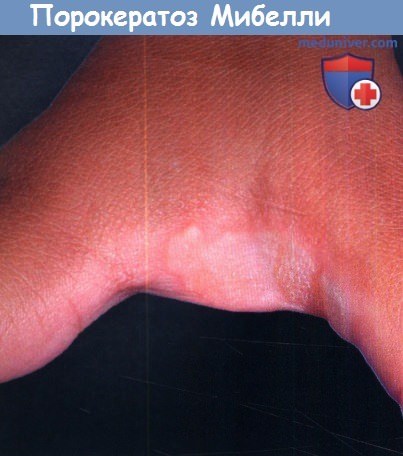
В данном случае из-за межпальцевой локализации шелушение по краю очага выражено слабо.
б) Диссеминированный поверхностный актинический порокератоз (ДПАП). ДПАП является наиболее распространенным типом порокератоза. Характерные очаги имеют форму однородных кольцевидных бессимптомных или сопровождающихся умеренным зудом папул диаметром от 2 до 5 мм, симметрично расположенных на конечностях. По сравнению с другими формами порокератоза высыпания при ДПАП носят более генерализованный характер.
В типичных случаях наблюдается более 50 очагов, локализованных преимущественно на участках кожи, открытых воздействию солнечных лучей. Несмотря на обширный характер высыпаний, поражение, как правило, не распространяется на ладони, подошвы и слизистые оболочки. По сравнению с порокератозом Мибелли, при ДПАП гиперкератотический край едва различим.
По мере того как очаги прогрессируют, их центральные области становятся атрофичными и ангидротическими. В большинстве случаев ДПАП наследуется по аутосомно-доминантному типу. Самый ранний возраст начала заболевания среди известных случаев—7 лет. Полная пенетрантность наблюдается не позднее 30-40 лет. Результаты первоначальных исследований, согласно которым очаги ДПАП развиваются под воздействием ультрафиолетового излучения, а фибробласты, полученные из кожи пациентов с ДПАП, демонстрируют гиперчувствительность к рентгеновским лучам, в дальнейшем последовательно воспроизвести не удалось; в связи с этим патогенез ДПАП до сих пор остается невыясненным.
в) Диссеминированный поверхностный порокератоз (ДПП). ДПП, как и ДПАП, наследуется по аутосомно-доми-нантному типу и развивается в возрасте 30-40 лет. Первичные очаги ДПП морфологически идентичны очагам ДПАП и в типичных случаях симметрично распределяются на конечностях; однако в отличие от ДПАП при ДПП поражение не распространяется на участки кожи, открытые воздействию солнечных лучей. Как и при ДПАП, при ДПП может наблюдаться свыше 100 очагов, локализованных преимущественно на разгибательных поверхностях конечностей. Необходимо особо отметить, что для обоих типов порокератоза нехарактерно поражение лица. И ДПАП, и ДПП чаще встречаются у женщин: соотношение женщин и мужчин, страдающих данными формами порокератоза, составляет 3:1.
г) Диссеминированный поверхностый порокератоз при иммуносупрессии (ДППИ). Диссеминированный поверхностный порокератоз при иммуносупрессии (ДППИ) развивается у следующих групп лиц: у пациентов, перенесших трансплантацию почек, печени или сердца; у лиц, подвергавшихся облучению; у больных, проходивших курс иммуносупрессивной химиотерапии или терапии системными кортикостероидами; у пациентов со злокачественными заболеваниями крови; у лиц с инфекцией вируса иммунодефицита человека.
Случаи порокератоза зафиксированы также у лиц, перенесших трансплантацию костного мозга при отсутствии непрерывной иммуносупрессивной терапии. Данный факт свидетельствует о том, что иммуносупрессия не является единственной ассоциацией при ДППИ. Характер распространения и морфология очагов при ДППИ аналогичны ДПАП, однако в случае ДППИ связь с воздействием солнечных лучей менее очевидна.
д) Линейный порокератоз. Линейный порокератоз — необычный вариант, традиционно рассматриваемый как отдельный синдром, но все более и более признаваемый как мозаичное проявление одного из других типов порокератоза. Как правило, заболевание возникает в раннем детском возрасте, хотя также зафиксированы врожденные случаи. Описаны два варианта линейного порокератоза. В первом из них клиническая картина представлена односторонним поражением конечности, распространяющимся вдоль линий Блашко. Реже наблюдается второй—генерализованный — вариант линейного порокератоза, при котором множественные очаги поражают несколько конечностей и могут распространяться на туловище.
По сравнению с другими разновидностями заболевания линейный тип порокератоза обладает наиболее высоким злокачественным потенциалом, что предположительно связано с утратой аллели вследствие мутации. У пациента в оригинальной публикации Мибелли, вероятнее всего, имелся одновременно линейный порокератоз и ДПАП, этот феномен был подтвержден несколькими последующими сообщениями. Данный факт, служащий примером сегментных манифестаций типа 2 аутосомно-доминантного заболевания, может объясняться потерей гетерозиготности аллели при ДПАП.
е) Диссеминированный порокератоз ладоней и подошв (ДПЛП). Диссеминированный порокератоз ладоней и подошв (ДПЛП) (точечный порокератоз ладоней и подошв) представляет собой генодерматоз аутосомно-доминантного типа наследования. Для заболевания характерны небольшие, относительно однородные очаги, первоначально возникающие на ладонях и подошвах. Впоследствии поражение распространяется на другие участки тела, включая слизистые оболочки и зоны, не подверженные открытому воздействию солнца. Очаги на ладонях и подошвах, как правило, носят более выраженный гиперкератотический характер, а специфические продольные бороздки, проходящие вдоль этих выступов, нередко принимают достаточно отчетливую форму.
В типичных случаях очаги развиваются в подростковом или юношеском возрасте, для них характерно двустороннее симметричное распределение. ДПЛП встречается у мужчин в два раза чаще, чем у женщин.
ж) Точечный порокератоз. Точечный порокератоз в типичных случаях проявляется в подростковом или юношеском возрасте. Заболевание может сочетаться с другими типами порокератоза. Клиническая картина представлена многочисленными мелкими разрозненными точечными гиперкератотическими очагами на ладонях и подошвах, окруженными тонкими приподнятыми краями. Очаги могут располагаться линейно либо сливаться, образуя бляшки. Точечный порокератоз необходимо клинически и гистологически дифференцировать от точечной кератодермии, также известной как точечная порокератотическая кератодермия или точечный порокератоз ладоней и подошв.
з) КЗАУК-синдром. КЗАУК-синдром (краниосиностоз и ключичная гипоплазия, задержка закрытия родничка, анальные аномалии, урогенитальные пороки развития, кожные высыпания), также известный как КАП-синдром (краниосиностоз, анальные аномалии, порокератоз), является редким генодерматозом. К настоящему моменту заболевание зафиксировано лишь в четырех этнически различных семьях. Основные фенотипические признаки синдрома включают краниосиностоз с гипоплазией ключиц, аномалии ануса и порокератоз. Заболевание наследуется по аутосомно-рецессивному типу и, возможно, связано с хромосомной полосой 22q12-13. Кожные проявления синдрома отличаются постоянством.
Начиная с месячного возраста у больных развивается обширное поражение, представленное маленькими порокератотическими папулами, локализованными преимущественно на лице и конечностях. Зафиксированы обострения очагов вследствие воздействия солнечных лучей. Предполагается, что причиной заболевания является нарушение регуляции многочисленных сигнальных механизмов во время эмбриогенеза.

Отчетливо видны выступы и бороздки.
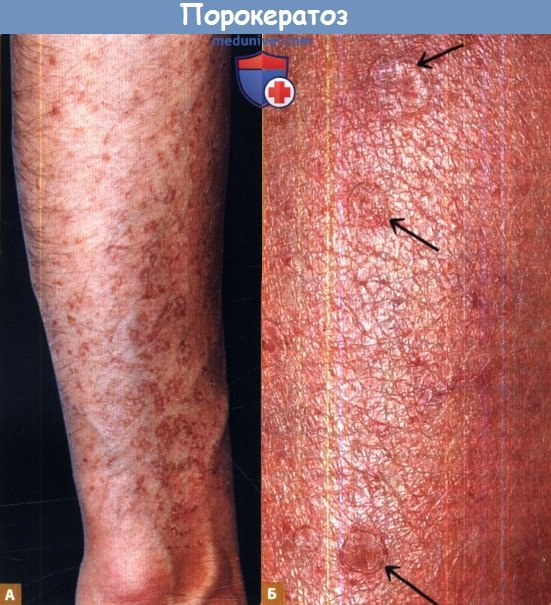
Б. У этого пациента на предплечье отчетливо видны характерные бороздки (показаны стрелками).
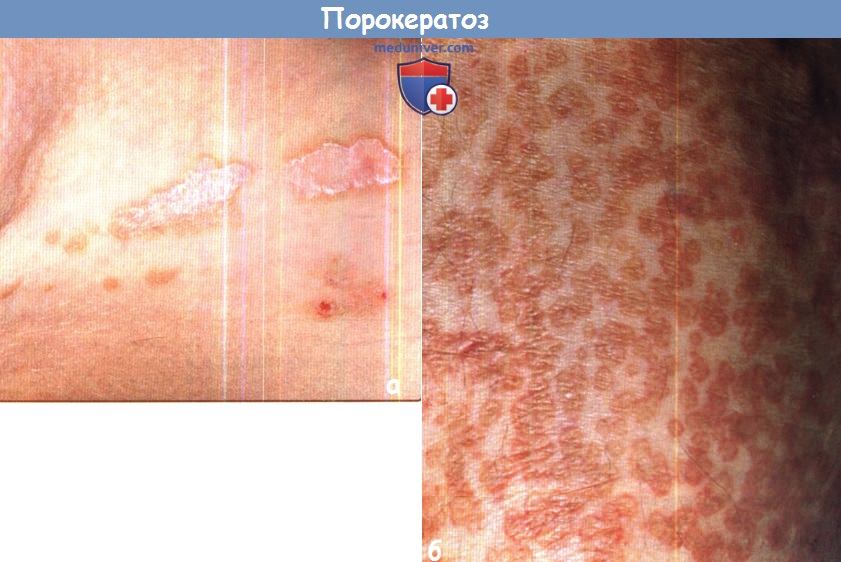
б - Диссеминированный порокератоз ладоней и подошв с многочисленными поверхностными очагами в икроножной области.
Обратите внимание на сходство с картиной диссеминированного поверхностного актинического порокератоза на рисунке выше.
- Рекомендуем далее ознакомиться со статьей "Гистология и дифференциация порокератоза"
Оглавление темы "Порокератоз.":