
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизмы участия эндотелия в воспалении
а) Эндотелий и врожденный иммунитет. Врожденный иммунитет, также называемый естественным или нативным, обеспечивает защиту хозяина от микроорганизмов, которая не зависит от Т-и В-лимфоцитов и их рецепторов к специфическим антигенам. Система врожденного иммунитета эволюционно значительно древнее, чем Т- и В-клеточная специфическая иммунная система (часто называемая адаптивным иммунитетом, чтобы отличать его от врожденного). Гуморальный компонент прежде всего включает систему комплемента. Основные клеточные эффекторы врожденного иммунитета у человека — нейтрофилы, мононуклеарные фагоциты и естественные киллеры, однако в нем также участвуют эндотелиальные клетки (ЭК).
Эндотелиальные клетки являются первоначальной целью при различных типах инфекций. Например, под действием липополисахарида — основного компонента клеточной стенки грамотрицательных бактерий, эндотелий активируется рецепторным комплексом, состоящим из TLR-4, CD14 и MD2. Затем присоединение адапторного белка — миелоидного фактора дифференциации (MyD88) к лиганд-активированному TLR-4 рецептору запускает МуD88-зависимый механизм, который приводит к ранней активации ядерного фактора каппа-В и митоген-активируемых протеинкиназ. Одновременно с этим, независимые от MyD88 механизмы, используя альтернативный адаптерный белок TRIF, приводят к поздней активации ядерного фактора -κВ, а также к производству интерферонов. Эндотелиальные клетки (ЭК) также экспрессируют TLR-2, который служит сигнальным рецептором для компонентов клеточной стенки грам-положительных бактерий; при вирусных инфекциях обычно задействованы внутриклеточные TLR, которые связываются с различными нуклеиновыми кислотами. Стимуляция TLR2/6 бактериальными липопептидами также может способствовать ангиогенезу и приводит к секреции колониестимулирующего фактора гранулоцитов и макрофагов (GM-CSF). В определенных сосудистых руслах экспрессируется макрофагальный рецептор маннозы (CD206) — рецептор к олигосахаридам на клеточной стенке бактерий, дрожжей и паразитов. Другие рецепторы врожденной иммунной системы — различные лектин-подобные молекулы, например, связывающий маннозу белок или галактины, которые распознают бактериальные углеводы. Белок, связывающий маннозу, может стимулировать альтернативные пути активации комплемента.
TNF и II-1 — центральные эффекторные цитокины врожденного иммунного ответа. В целом эндотелиальный ответ на TNF и IL-1 схожи и рассматриваются совместно. Основной изоформой IL-1, которая синтезируется в ходе реакций врожденного иммунитета, является изоформа IL-β, а поскольку IL-α, основная изоформа, производимая эпителиальными клетками, по своим действиям неотличима, мы будем обозначать обе изоформы просто как IL-1. Изучены не менее пяти изменений фенотипа ЭК, которые обеспечивают воспаление и обобщенно называются активацией эндотелия.
1. Синтез вазодилататоров. Под действием TNF и IL-1 в ЭК повышается продукция таких вазодилятаторов, как PgI2, и NO. Они расслабляют гладкие мышцы, вызывая вазодилятацию и увеличение перфузии ткани. PgI2, также называемый простациклин, синтезируется в ЭК в ходе трех последовательных ферментативных реакций. ЭК экспрессируют одну из изоформ простагландин Н-синтазы (PGHS-1), также называемую циклооксигеназой 1, которая проявляет конститутивную активность. Под действием TNF и II-1 в ЭК значительно повышается активность PGHS за счет индукции экспрессии de novo второй изоформы, PGHS-2, также называемой циклооксигеназой 2. Этот ответ значительно повышает способность производить из арахидоно-вой кислоты простагландин Н2 и впоследствии PGI,. Считается, что фармакологическая редукция образования PGI2 вносит вклад в противовоспалительное действие аспирина и нестероидных противовоспалительных препаратов, которые ингибируют активность PGHS. NO образуется при превращении аргинина в цитруллин при помощи eNOS. TNF оказывает противоположные действия на вазодилатацию. Вначале он усиливает функцию eNOS и синтез NO, активируя Akt и повышая синтез тетрагидробиоптерина, кофактора, ограничивающего функцию eNOS. Позднее TNF дестабилизирует мРНК синтазы eNOS, ингибируя таким образом синтез NO.
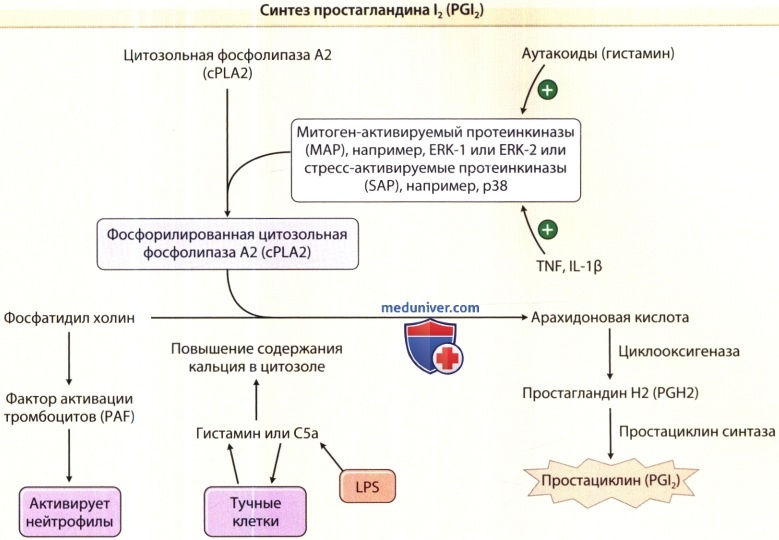
ERK — внеклеточная сигнал-регулируемая киназа; IL — интерлейкин; LPS — липополисахарид; TNF — фактор некроза опухоли.
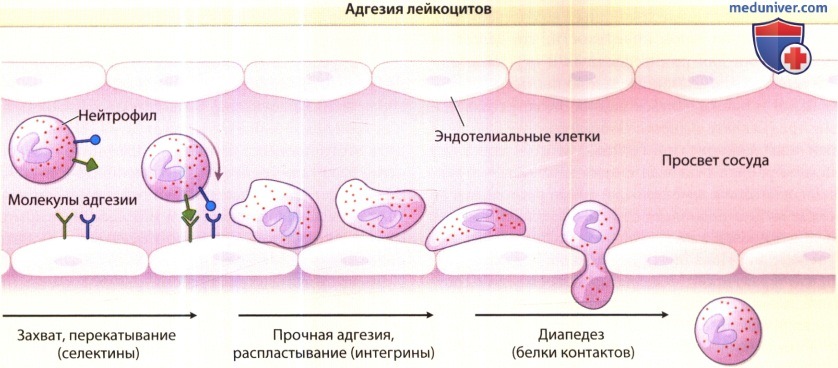
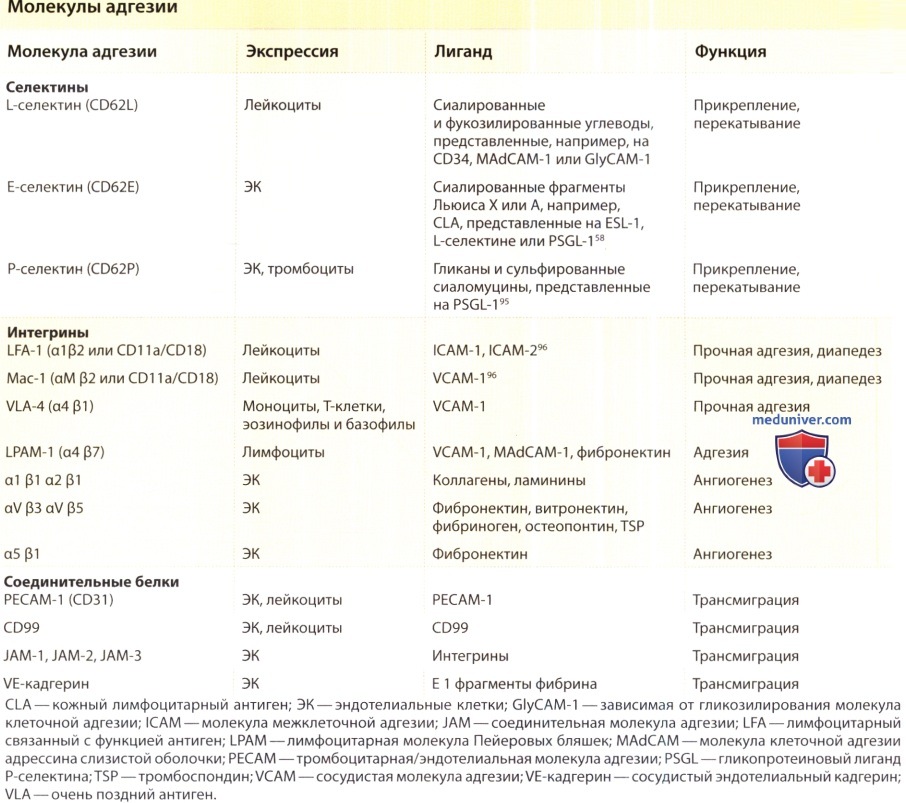
2. Адгезия лейкоцитов. Под действием TNF и IL-1 у эндотелиальных клеток (ЭК) меняется поверхность для облегчения инициального прикрепления, перекатывания и, наконец, прочной адгезии и распространения лейкоцитов. Адгезия всегда предшествует выходу лейкоцитов в ткани из сосудов. Прикрепление и перекатывание обычно обеспечивают селектины, экспрессирующиеся на лейкоцитах или ЭК. Существует три структурно родственных белка селектина; (1) L-селектин (CD62L), (2) Е-селектин (CD62E) и (3) Р-селектин (CD62P). При наличии кровотока лейкоцит вращается в направлении кровотока, быстро разрывая и образуя заново селектиновые соединения. Это приводит к перекатыванию лейкоцита вдоль поверхности ЭК. Лиганды, участвующие в селектинопосредованной адгезии и инициальном прикреплении, могут отличаться от участвующих в перекатывании; например, во время этих двух фаз взаимодействия нейтрофилы могут переключаться с лиганда-1 L-селектина на лиганд-1 Е-селектин как основной лиганд Е-селектина. В исследованиях с использованием блокирующих моноклональных антител или мышей с нокаутированными молекулами адгезии установлено, что in vivo селектины требуются для перекатывания и последующего привлечения лейкоцитов, несмотря на сохранение функции Е- и Р-селектинов (для полного прекращения перекатывания должны быть нокаутированы или ингибированы обе молекулы). Следует отметить, что у мышей, IL-1 и TNF стимулируют синтез как Р-, так и Е-селектина, но у людей они индуцируют синтез только Е-, но не Р-селектина.
Экспрессия селектина Е у людей во многом зависит от индуцируемого цитокинами синтеза de novo, в то время как Р-селектин предварительно формируется и мобилизуется на клеточную поверхность из ТВП в ответ на такие стимулы как тромбин, гистамин, реактивные виды кислорода и т.п. Следовательно, Е-и Р- селектин у человека не дублируются. Экспрессия селектина происходит быстро: Р-селектина в течение 5 минут и Е-селектина в течение 1-2 часов, но она часто транзиторная и достигает пика через 15 минут у Р-селектина и через 4-5 часов у Е-селектина, хотя может персистировать гораздо дольше на эпителиальных клетках кожи. Еще одним важным механизмом, инициирующим перекатывание Т-лимфоцитов на ЭК, является взаимодействие рецептора гиалуронана CD44. CD44 экспрессирован на эндотелиальных клетках и в случае воспаления связывается с гиалуронаном, который затем связывается с CD44, экспрессированном на активированных Т- лимфоцитах и макрофагах.
Прочная адгезия лейкоцитов к ЭК в основном обеспечивается интегринами лейкоцитов. Для взаимодействия с ЭК лейкоциты используют два вида интегринов. Первый из них — CD18 или β2 интегрины, в том числе функционально связанный с лейкоцитами антиген-1 (αLβ2 или CD11a/CD18) и Мас-1 (αMβ2 или CD11b/CD18). Адгезия, обеспеченная узнаванием LFA-1 или Мас-1 межклеточной молекулы адгезии 1 (ICAM-1) значительно сильнее, чем обеспеченная селектинами, поэтому если эти молекулы занимают ICAM-1, лейкоциты больше не перекатываются. В случае нейтрофилов участие интегринов приводит к постепенному замедлению скорости качения. Напротив, лимфоциты переходят от перекатывания к прочной адгезии. После адгезии лейкоциты распластываются и медленно ползут вдоль поверхности ЭК до клеточного контакта, где происходит диапедез через стенку сосуда в ткани. Вероятно, в распластывании, переползании и диапедезе лейкоцитов играет роль прикрепление при помощи LFA-1, отсоединение и повторное прикрепление к ICAM-1. Несмотря на то что вначале (в первые четыре часа после обработки TNF) количество ICAM-1 на люминальной поверхности ЭК повышается, в первые несколько дней он локализуется преимущественно в клеточных контактах. В исследованиях с блокирующими антителами у нокаутных мышей выявлено, что решающую роль в адгезии лейкоцитов к ЭК и последующем привлечении лейкоцитов играют β, интегрины и ICAM-1.
Вторая группа лейкоцитарных интегринов, обеспечивающих прикрепление к ЭК,— α4 интегрины (CD49d), образующие комплекс либо с β1 (CD29) с формированием очень позднего антигена-4 (VLA-4) (α4β1), либо с β7 с формированием лимфоцитарной молекулы адгезии пейеровых бляшек (интегрин α4β7). Основной лиганд ЭК для VLA-4 — сосудистая молекула адгезии 1 (VCAM-1). Этот представитель надсемейства иммуноглобулинов не экспрессируется на ЭК в покое при обычных условиях, однако увеличивается под действием TNF или IL-1 в течение некоторого периода времени и достигает пика через 24 часа. Количество VCAM-1 может селективно увеличиваться IL-4 цитокином, связанным с определенными видами адаптивного, а не врожденного иммунитета. Экспрессия самого VCAM-1 не отвечает на интерферон-γ (IFN-γ), однако IFN усиливает вызванную TNF экспрессию VCAM-1, Интересно, что VCAM-1 может стимулировать начальное прикрепление и перекатывание некоторых лейкоцитов (например, Т-клеток, но не нейтрофилов), а также прочную адгезию в зависимости от того, повышается ли аффинность/авидность лейкоцитарного VLA-4 при активации лейкоцита. В экспериментах с блокирующими антителами подтверждена роль этих молекул в привлечении лейкоцитов in vivo. Мыши, нокаутные именно по VCAM-1 гибнут на стадии эмбриона из-за недостаточной плацентации. У некоторых нокаутных по VCAM-1 мышей был разрушен первый экзон. У таких животных синтезировался укороченный белок VCAM-1, у которого отсутствовал первый Ig-домен, но они экспрессировали его на пониженных («гипофморфных») уровнях. Эти животные также подтверждают роль VCAM-1 в рекрутировании лейкоцитов.
3. Активация лейкоцитов. ЭК под действием TNF или IL-1 вызывает активацию прикрепленных или перекатывающихся лейкоцитов, запуская переход от обусловленного селектинами низкоаффинного прикрепления к обусловленной интегринами прочной адгезии и переход от сферических неподвижных лейкоцитов к распространяющимся мигрирующим формам. В некоторых экспериментах, по-видимому, для запуска этих изменений достаточно участия селектинов, однако в большинстве работ было показано существование определенного сигнала. Один из кандидатов в активаторы — липидный медиатор — фактор активации тромбоцитов (PAF), который может быть расположен на плазматической мембране ЭК. Синтез PAF инициируется активацией цитозольной фосфолипазы А2, то есть той же реакцией, в которой образуется арахидоновая кислота для синтеза. Сообщали, что IL-1β может стимулировать синтез PAF в ЭК, однако в большинстве исследований подчеркивают роль аутакоидов (например, гистамина), но не цитокинов в качестве индукторов синтеза PAF.
Хемокины — важная группа TNF или IL-1-индуцируемых активаторов лейкоцитов, для которых характерно различное количество аминокислот между двумя цистеиновыми остатками на N-конце (то есть СС, СХС и т.д.). Хемокины вызывают миграцию и/или активацию различных типов лейкоцитов при помощи взаимодействия группы из семи трансмембранных связанных с G-белками рецепторов. СХС хемокины (например, IL-8) особенно важны для активации нейтрофилов, тогда как СС хемокины (например, МСР-1) важны для активации моноцитов. IL-8 хранится в ТВП и высвобождается при стимуляции гистамином или тромбином. Под действием TNF или IL-1 β ЭК синтезируют и выделяют IL-8 или МСР-1. Помимо этого, ЭК могут предъявлять выделяемые хемокины в комплексе с протеогликанами клеточной поверхности, что делает их биодоступными для прикрепленных и перекатывающихся лейкоцитов. Связанные хемокины, представленные ЭК, могут производиться ЭК, или ЭК могут захватывать и предъявлять вещества, образованные клетками тканей или лейкоцитами. Помимо этого, существует трансмембранная белковая форма хемокина — фракталкин, в котором хемокиновый домен расположен на вершине муцин-подобного стебля. Фракталкин может быть индуцирован в ЭК сосудов, и он действует как молекула адгезии. Появляется все больше доказательств того, что фракталкин — медиатор повреждения сосудов при гломерулонефрите, отторжении аллотрансплантата и атеросклерозе. Хемокины также могут играть роль в активации и привлечении определенных подгрупп лейкоцитов, как обсуждается далее.
4. Экстравазация лейкоцитов. Для осуществления эстравазации лейкоцитов необходимы предшествующие этапы прикрепления, перекатывания и адгезии, за которыми следует их перемещение к межклеточным контактам. Белки межклеточных контактов, такие как РЕСАМ-1 (CD31), CD99 и адгезионные молекулы этих контактов (JAMs) способствуют трансмиграции лейкоцитов, а активированные ЭК, как отмечалось ранее, накапливают в этих же межклеточных контактах адгезивные лиганды, такие как Е-селектин, ICAM-1 и VCAM-1. Детали взаимодействия между лейкоцитами и ЭК во время диапедеза in vivo не вполне выяснены. По-видимому, основной путь экстравазации — межклеточные контакты. Антитела к РЕСАМ-1 ингибируют этот процесс. Следует отметить, что у мышей с нокаутной РЕСАМ-1 влияние на трансмиграцию лейкоцитов зависит от линии мышей. РЕСАМ-1 у границы эндотелия формирует гомофильное взаимодействие между РЕСАМ-1 на лейкоцитах и запускает целевую рециркуляцию мембраны компартмента, локализованного в ретикулуме рядом с латеральной границей эндотелиальной клетки, который называется латеральным пограничным компартментом рециркуляции. Цикл этого компартмента конституционально возобновляется и, мобилизованный к участкам межклеточных контактов, он окружает трансмигрирующие лейкоциты. Этот компартмент содержит также CD99 и JAM-1, но не VE-кадгерин. Белки JАМ взаимодействуют с лейкоцитами, связываясь с интегринами.
Интегрин LFA-1 обеспечивает адгезию нейтрофилов и Т-клеток к JAM-A; интегрин α4β1 обеспечивает адгезию лейкоцита к JAM-B. Взаимодействия JAM-2- α4β15 по-видимому, зависят от взаимодействия между JAM-B и JAM-B. JAM-B связывается с CD11b/CD18 на нейтрофилах, и считается, что это взаимодействие важно для трансэпителиальной миграции нейтрофилов. Было показано, что для трансмиграции лейкоцитов необходимо формирование VE-кадгеринами межклеточных щелей и что р120 является важным внутриклеточным медиатором этого процесса, поскольку регулирует экспрессию VE-кадгерина в межклеточных контактах. Кроме того, VE-кадгерин может взаимодействовать с лейкоцитами через промежуточный фрагмент фибрина, который образуется благодаря индуцируемой тромбином активации фибриногена и последующей переработке плазмином или урокиназой.
В определенных обстоятельствах лейкоциты могут выходить из сосудов через тело ЭК. Факторы, которые влияют на выбор лейкоцитами межклеточного или трансклеточного пути, неизвестны. На ранних стадиях этого процесса происходит фагоцитоз лейкоцита через выступы мембраны, богатые ICAM-1 и VCAM-1. В трансклеточном диапедезе моноцитов и нейтрофилов участвует также латеральный пограничный компартмент рециркуляции, в котором важнейшими адгезивными молекулами являются РЕСАМ-1, CD99 и JAM-А. Помимо этого, быстрая трансмиграция Т-клеток (но не других групп лейкоцитов) зависит от наличия венулярного уровня нагрузки смещения, который посылает сигнал Т-клеткам, «растягивая» прикрепленные адгезивные молекулы, такие как LFA-1. Под действием TNF или IL-1 ЭК секретируют и активируют матриксные металлопротеиназы (MMPs), которые разрушают плотный участок матрикса соединительной ткани, то есть базальную мембрану, под эндотелиальным монослоем. ЭК также могут стимулировать синтез или высвобождение ММР, и сигналом для этого высвобождения могут быть цитокин-индуцибельные молекулы, например, VCAM или хемокины.
Циркулирующие в крови Т-клетки являются гетерогенными. Некоторые из них никогда не встречались с родственным антигеном и считаются наивными. Т-клетки памяти возникают после встреч с антигеном и подразделяются на субпопуляции центральных и эффекторных клеток. Т- клетки из субпопуляции эффекторных клеток памяти типа CD45RO+CCR7low L-силектин lowCD4+ трансмигрируют через монослои эндотелия в камеры потока в присутствии индуцируемого интерфероном-γ белка -10 (IP-10) (CXCL10), в то время как трансмиграции наивных клеток или Т-клеток памяти центральной популяции не происходит, несмотря на экспрессию CXCR3 на клетках этого типа. Путем анализа in vitro установлено, что распознавание Т-клетками эндотелиальных молекул МНС класса II блокирует реакцию на IP-10, но вызывает отсроченную трансэндотелиальную миграцию эффекторных Т-клеток памяти, но не наивных или центральных Т-клеток памяти CD4(+). Фракталкин, синтезируемый на ЭК микрососудов под действием TNF или IL-1, также способствует осуществлению реакции, инициируемой Т-клеточными рецепторами (TCR).70 В механизме действия TCR участвует также молекула РЕСАМ-1, которая не используется Т- клетками, реагирующими на IP-10.
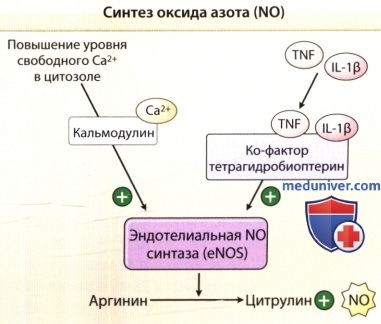
IL — интерлейкин, TNF — фактор некроза опухоли.
5. Экстравазация и провизионный матрикс. Экстравазация белков плазмы в ткани — признак воспаления. Антакоиды, например, гистамин, позволяют этому процессу происходить быстро и транзиторно, вызывая сокращение актинового цитоскелета ЭК, что открывает промежутки между клетками. VEGF может действовать сходным образом, однако также оказывает основные эффекты на транспорт жидкости через везикуло-вакуолярные органеллы. Напротив, под действием TNF или IL-1 ЭК допускают неспецифическую экстравазацию жидкости и белков плазмы только через несколько часов через процесс, зависящий от синтеза новых белков. Утрата селективности — признак воспаления. Основная цель этой реакции — создание в тканях отложения белков плазмы, в частности, фибронектина и фибриногена, которые образуют провизионный матрикс для подвижных лейкоцитов. Это важно, поскольку у циркулирующих лейкоцитов обычно нет рецепторов для взаимодействия с нормальным внеклеточным матриксом, состоящим в основном из интерстициальных коллагенов.
В сочетании эти пять опосредованных TNF или IL-1 ответов ЭК обеспечивают развитие воспалительного инфильтрата, исходно состоящего из нейтрофилов, а затем из мононуклеарных фагоцитов. Инфильтрирующие лейкоциты служат для фагоцитоза обнаруженных микроорганизмов и удаления поврежденных тканей. Инфильтрирующие фагоциты могут повреждать ткани за счет действия тех же медиаторов и ферментов, которые используются для устранения микроорганизмов. Эти процессы могут и часто повреждают ЭК, что проявляется ретракцией ЭК и/или денудацией, а также часто сопровождается внутрисосудистым тромбозом. Внутрисосудистые тромбы вызывают ишемию и повреждение тканей. К счастью, под действием TNF и IL-1 ЭК становятся более резистентными к повреждению за счет экспрессии протективных продуктов генов, в частности, антидотов активных форм кислорода (например, марганец, супероксиддисмутазы и гемоксигеназы), ингибиторов протеаз (например, ингибитора активатора празминогена) и тканевых ингибиторов металлопротеиназ, а также цитопротективных генов (белка цинковых пальцев А20) и родственных Bel антиапоптотических генов (например, А1). Эти протективные реакции позволяют ЭК перенести многие воспалительные реакции без запуска тромбоза и инфаркта тканей.
Приобретение цитопротективных черт наблюдалось в ЭК трансплантатов, которые избежали отторжения. Тем не менее этих протективных реакций не всегда достаточно. Более того, некоторые другие обусловленные TNF и IL-1 ответы эндотелия, например, индукция прокоагулянтных белков (в том числе тканевых факторов), а также утрата антикоагулянтных белков (в том числе тромбомодулина), могут в действительности вызывать повреждение тканей и сосудов. Это сочетание привлечения и активации нейтрофилов с изменениями в эндотелиальной системе коагуляции может быть причиной повреждения ткани при местной реакции Шварцмана или грам-отрицательной септицемии.
Итак, ЭК при помощи ответов на цитокины могут вносить вклад в защитные реакции хозяина за счет врожденной иммунной системы. Тем не менее те же эффекторные механизмы могут повреждать эндотелий и ткани.
б) Роль эндотелия в адаптивном иммунитете. Эндотелиальные клетки (ЭК) микрососудов конститутивно экспрессируют молекулы МНС. К настоящему времени единственная известная функция молекул МНС класса I и II — презентация пептидов Т-клеткам. Эндотелиальные клетки (ЭК) сосудов не только экспрессируют МНС, но и белки, задействованные в образовании пептидов и переносе этих пептидов в образующиеся молекулы класса I и II. Экспрессия генов класса I (а также генов, кодирующих ассоциированную молекулу β2-микроглобулина) и белков, участвующих в образовании пептидов и присоединение пептида к молекуле МНС класса I, усиливаются цитокинами, особенно интерферонами и TNF. В стандартных условиях культивирования ЭК человека не экспрессируют молекулы МНС класса II или белки, отвечающие за присоединение пептидов к молекулам класса II, однако эти белки индуцируются IFN-γ. У человека молекулы класса II постоянно экспрессируются в клетках микрососудов in vivo. До сих пор неясно, зависит ли эта «конститутивная» экспрессия от низкого уровня циркулирующего IFN-γ или является истинно конститутивной.
Типичный диаметр просвета капилляров составляет 10 мкм, то есть меньше, чем диаметр циркулирующего в крови лимфоцита. Следовательно, циркулирующие Т-клетки вынуждены контактировать с антигенными пептидами в комплексе с молекулами МНС класса I и II, представленными на поверхности эндотелия при нормальной циркуляции. Основной вопрос состоит в том, какое действие этот контакт оказывает на Т-клетки. В исследованиях последствий узнавания Т-клетками антигенов, презентированных ЭК, в основном применялись клеточные культуры. Наивные Т-клетки игнорируют антигены, презентированные ЭК, тогда как покоящиеся Т-клетки памяти или недавно активированные Т-бласты отвечали экспрессией генов цитокинов и в некоторой степени пролиферацией. Это ограничение согласуется с данными о том, что ЭК человека не в полной мере экспрессируют определенные ко-стимулирующие молекулы, например, CD80 и CD86, и в основном ко-стимулируют Т-клетки LFA-3 (CD58). Наивные Т-клетки мало экспрессируют CD2 — рецептор для LFA-3. Другой фактор состоит в том, что наивные Т-клетки недостаточно экспрессируют молекулы адгезии, необходимые для прочного прикрепления к ЭК. Помимо конститутивной экспрессии LFA-3, в ЭК человека происходит индуцибельная экспрессия лигандов для индуцибельных ко-стимуляторов Т-клеток — CD137 и CD134, которые обычно связаны со стимуляцией или реактивацией Т-клеток памяти, а также экспрессией молекул, таких как лиганды для PD-1, которые ингибируют активацию Т-клеток.
Предполагали, что презентация антигена ЭК in vivo выполняет несколько не взаимоисключающих функций. Первая из них—поддержание иммунологической памяти. Долгоживущие Т-клетки памяти могут периодически нуждаться в низкоаффинном сигнале, чтобы выжить, и экспрессия молекул МНС, несущих собственные пептиды ЭК, может обеспечить такие низкоаффинные сигналы, поскольку эти клетки проходят через микроциркуляторное русло. Недавно полученные данные показывают, что ЭК человеческой крови способны презентировать аутоантигены in vivo, что возможно помогает поддерживать жизнеспособность популяции клеток памяти. Вторая функция может приобретать значение при повторном инфицировании определенным патогеном. В данном случае ЭК могут презентировать пептиды микробного происхождения, связанные с молекулами МНС, что обеспечивает антиген-специфичный сигнал циркулирующим Т-клеткам о присутствии патогена. При наличии этого механизма он служит для повышения иммунного контроля. Это потенциально важная функция, поскольку Т-клетки, специфичные к конкретному антигену, образовавшиеся при контакте с антигеном, редки (менее 1 на 10000-100000). Презентация антигена ЭК может значительно повысить вероятность того, что антиген будет вовремя распознан, и что Т-клетки памяти будут привлечены в место угрозы. Третья возможная функция — пост-тимическая индукция и активация CD4+25+ белка Р3 регуляторных Т-клеток.
Показано, что у мышей аллоантигенная презентация сосудистыми ЭК CD4+ Т-клеткам индуцирует активность регуляторных Т-клеток, которые могут быть необходимы для развития толерантности. Тем не менее следует отметить, что ЭК мыши обычно не экспрессируют молекулы МНС II in vivo и что Т-клетки человека в культуре преимущественно активируют CD4+ эффекторные клетки, а не регуляторные Т-клетки.
Т-клетки могут дифференцироваться в специализированные цитокин-продуцирующие подгруппы. У CD4+ Т-клеток такой ответ может быть поляризован либо как ответ Т-хелперов 1 (Тh1), в котором доминируют Т-клетки, продуцирующие IFN и лимфотоксин, либо как ответ Т-хелперов 2 (Тh2, они продуцируют IL-4 и IL-5). Впоследствии было выяснено, что некоторые эффекторные CD4+ Т-клетки могут селективно секретировать IL-17 (провоспалительный цитокин). Т-хелперные клетки, продуцирующие IL-17 (Тh17), через LFA-1 связываются с молекулой ICAM-1, которая экспрессирована на ЭК. IL-17 идентифицирован как важный провоспалительный медиатор при различных заболеваниях, в том числе атопическом дерматите и бронхиальной астме. В различных моделях, IL-17 был также способен непосредственно нарушать гематоэнцефалический барьер вследствие прямого воздействия клеток Тh17 на ЭК. Нарушение гематоэнцефалического барьера является важным этапом в развитии воспаления центральной нервной системы, что наблюдается при таких заболеваниях как рассеянный склероз. На HUVEC, IL-17 индуцирует экспрессию молекул адгезии, таких как ICAM-1, VCAM-1, Е-селектин, но потенция IL-17 значительно ниже, чем у TNF или IL-1. Еще одна группа специализированных клеток — регуляторные Т-клетки (Treg; CD4+/CD25+/FOXP3+), которые играют важную роль при воспалительных заболеваниях и неоплазиях. ЭК сосудов в опухолях экспрессируют специфическую адресную сигнатуру для направления Tregs в опухоль.
Цитокины, высвобождаемые Th1 или Th2, по-разному активируют ЭК. Например, IFN-γ в сочетании с лимфотоксином вызывает длительную экспрессию Е-селектина в качестве важного лиганда Th1 Т-клеток. Th1 (но не Th2) способны связываться с Р-селектином и Е-селектином, миграция ТЫ в воспалительно измененную кожу блокируются антителами к Р- и Е-селектину. Гликопротеиновый лиганд 1 Р-селектина действует как лиганд Р- и Е-селектина для Th1 клеток, что коррелирует с повышенной экспрессией α3-фукозилтрансфераз в Th1-клетках. Напротив, образованный Th2 IL-4 повышает экспрессию VCAM-1, тогда как подавляется Е-селектин — молекула поверхности эозинофилов, при этом, возможно, подавляется привлечение Th2-клеток. Другими словами, цитокины, вырабатываемые Т-клетками, могут модифицировать врожденный ответ ЭК таким образом, что привлечение лейкоцитов смещается в сторону привлечения специфических эффекторных клеток. И наконец вклад в селективный рекрутмент подгрупп Т-клеток может вносить дифференцированная экспрессия хемокиновых рецепторов. Например, CCR5 и в меньшей степени CXCR3 обнаруживаются преимущественно на Th2-клетках.
- Рекомендуем далее ознакомиться со статьей "Микрососуды кожи при воспалении"
Редактор: Искандер Милевски. Дата публикации: 18.2.2019