
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизмы развития (патогенез) острой миелоидной лейкемии
Острая миелоидная лейкемия — это опухоль, образующаяся из гемопоэтической клетки-предшественника в результате приобретенных онкогенных мутаций, нарушающих дифференцировку, что приводит к накоплению в костном мозге незрелых миелобластов. Остановка развития клеток миелоидной линии приводит к недостаточности костного мозга, связанной с анемией, тромбоцитопенией и нейтропенией.
Острая миелоидная лейкемия (ОМЛ) может возникнуть в любом возрасте, частота ее увеличивается с возрастом, достигая пика после 60 лет. В США ежегодно регистрируют 13 тыс. новых случаев острой миелоидной лейкемии (ОМЛ).
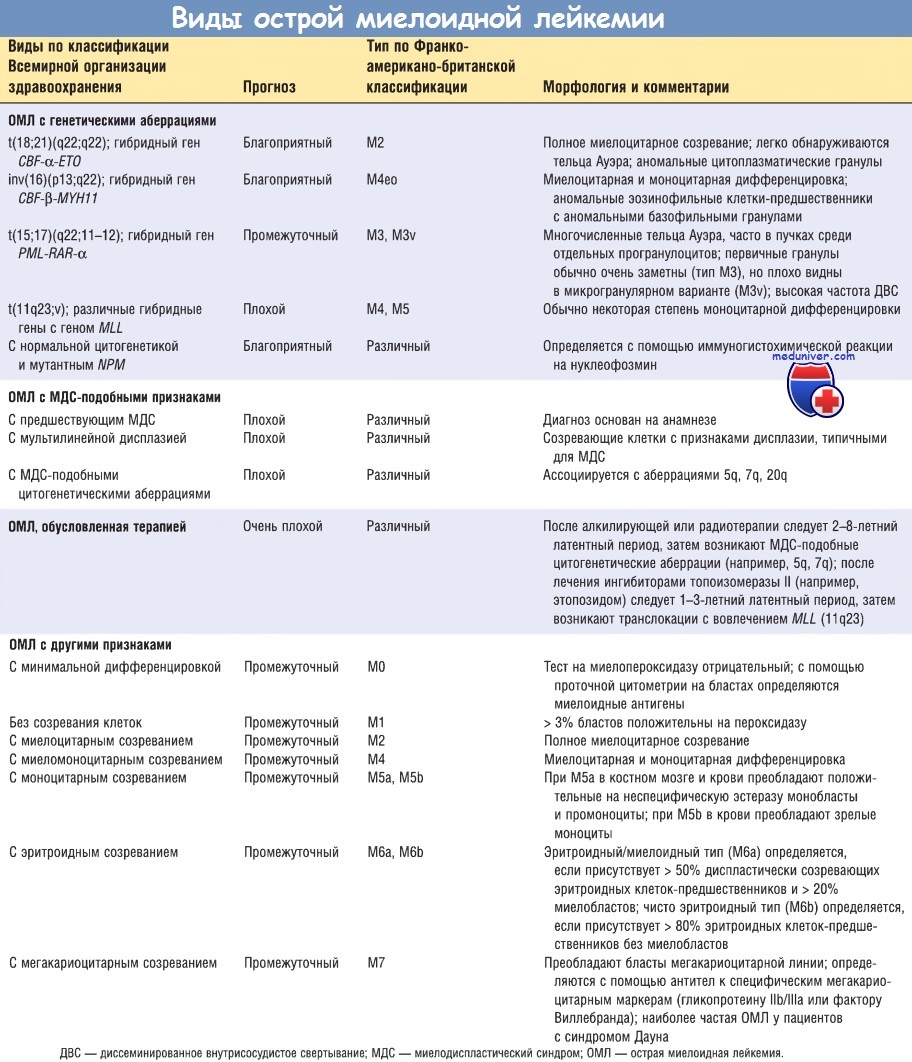
а) Классификация. Острая миелоидная лейкемия (ОМЛ) отличается крайней гетерогенностью, что отражает сложность дифференцировки миелоидных клеток. Новая классификация, предложенная ВОЗ, подразделяет ОМЛ на 4 вида. Первый из них включает формы ОМЛ, представляющие существенное значение, т.к. они коррелируют с прогнозом и определяют характер терапии.
Далее следуют виды ОМЛ, возникающие после миелодиспластических синдромов (МДС) или имеющие МДС-подобные признаки, и ОМЛ, обусловленные терапией. ОМЛ этих двух видов имеют отличительные генетические признаки и плохо поддаются лечению. Четвертый вид включает ОМЛ с другими признаками.
Острую миелоидную лейкемию (ОМЛ) классифицируют также по Франко-американо-британской классификации, в которой ОМЛ разделены на типы по степени дифференцировки и линиям лейкобластов. Применение этой классификации ограниченно, но ее еще используют на практике. В таблице ниже сопоставлены (насколько это возможно) классификация ВОЗ и Франко-американо-британской классификация. Учитывая возрастающую роль цитогенетических и молекулярных признаков в выборе терапии, необходимо продолжать дальнейшие разработки классификации ОМЛ.
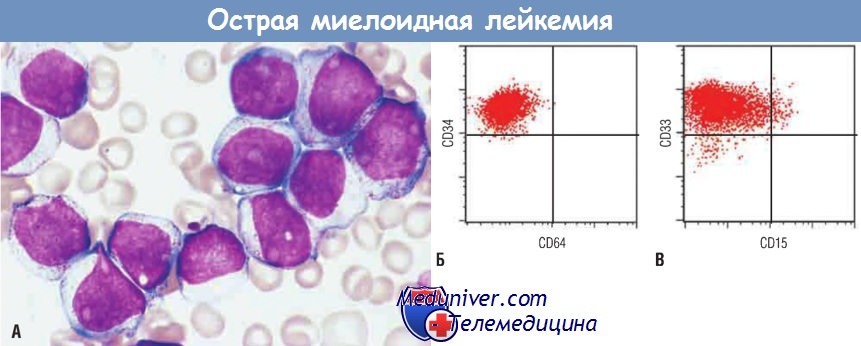
Миелобласты имеют тонкий ядерный хроматин, хорошо заметные ядрышки и мелкие азурофильные гранулы в цитоплазме.
(Б) Методом протонной цитометрии установлено, что миелобласты (изображены красными точками) экспрессируют CD34 (маркер мультипотентных стволовых клеток),
но не экспрессируют CD64 (маркер зрелых миелоидных клеток).
(В) Миелобласты экспрессируют CD33 (маркер незрелых миелоидных клеток),
а субпопуляция экспрессирует CD15 (маркер более зрелых миелоидных клеток).
Таким образом, эти бласты представляют собой миелоидные клетки с ограниченной степенью созревания.
б) Морфология острой миелоидной лейкемии (ОМЛ). Диагноз «острая миелоидная лейкемия» основан на наличии в костном мозге по меньшей мере 20% миелобластов. Различают несколько типов этих клеток. Опухоли могут содержать разные бласты или бласты со смешанными (гибридными) свойствами.
Миелобласты имеют тонкий ядерный хроматин, от 2 до 4 ядрышек и большее количество цитоплазмы по сравнению с лимфобластами. Цитоплазма часто содержит мелкие пероксидазоположительные азурофильные гранулы.
Во многих наблюдениях присутствуют тельца Ауэра (палочковидные включения) и отдельные, похожие на иглу азурофильные гранулы; они особенно многочисленны при ОМЛ с t(15;17) (острой промиелоцитарной лейкемии).
Монобласты имеют складчатое или дольчатое ядро, дают неспецифическую положительную реакцию на эстеразу и не содержат тельца Ауэра.
В некоторых случаях обнаруживается мегакариоцитарная дифференцировка бластов, часто сопровождающаяся фиброзом костного мозга, вызванным высвобождением фиброгенных цитокинов. В редких случаях бласты при ОМЛ несут признаки эритроидной дифференцировки.
Количество лейкозных клеток в крови сильно варьирует: от < 10 тыс. клеток/мм3 (50% пациентов) до > 100 тыс. клеток/мм3. Иногда бласты в крови полностью отсутствуют (алейкемическая лейкемия). По этой причине исследование костного мозга имеет большее значение и позволяет исключить острую лейкемию у пациентов с панцитопенией.
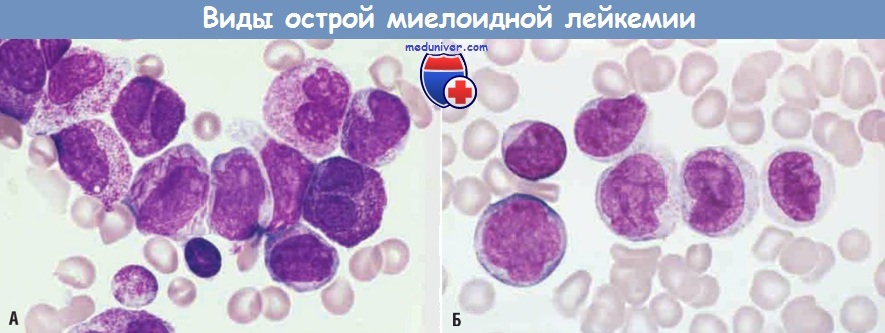
(А) Острая промиелоцитарная лейкемия с t(15;17) (тип М3 по Франко-американо-британской классификации).
В аспирате костного мозга обнаружены неопластические промиелоциты с многочисленными крупными аномальными азурофильными гранулами.
Другими характерными признаками являются присутствие клеток с двудольчатыми ядрами и клеток, содержащих многочисленные палочковидные включения (тельца Ауэра).
(Б) Острая миелоидная лейкемия с моноцитарной дифференцировкой (тип М5b по Франко-американо-британской классификации).
В мазке периферической крови видны монобласт и 5 промоноцитов со складчатой ядерной мембраной.
в) Иммунофенотип. Поскольку морфологически различить миелобласты и лимфобласты может быть трудно, диагноз «острая миелоидная лейкемия» подтверждают путем окрашивания на специфические миелоидные антигены.
г) Цитогенетика. Цитогенетический анализ играет основную роль в классификации ОМЛ. Кариотипические аберрации определяют в 50-70% случаев с помощью стандартных методов и в 90% случаев при использовании метода бэндинга с высоким разрешением. Отдельные хромосомные аномалии коррелируют с некоторыми клиническими признаками. ОМЛ, возникшая de novo у молодых взрослых, обычно ассоциируется со сбалансированными хромосомными перестройками, особенно t(8;21), inv(16) и t(15;17).
В отличие от этого при ОМЛ, возникающей после МДС либо воздействия ДНК-повреждающих агентов во время химиотерапии или радиотерапии, часто наблюдают моносомию или делеции, захватывающие 5-ю и 7-ю хромосомы, а хромосомные транслокации обычно отсутствуют. Исключение составляет ОМЛ, возникшая после лечения ингибиторами топоизомеразы II. Эта ОМЛ имеет сильную связь с транслокациями, включающими ген MLL на хромосоме 11q23. ОМЛ в пожилом возрасте с большей вероятностью ассоциируется с «плохими» аберрациями — делециями хромосом 5q и 7q.
д) Молекулярный патогенез. Многие повторные генетические аберрации, возникающие при ОМЛ, повреждают гены, кодирующие факторы транскрипции, необходимые для нормальной миелоидной дифференцировки. Так, две наиболее частые хромосомные перестройки, t(8;21) и inv(16), повреждают гены CBF-1 а и CBF-1b соответственно. Эти два гена кодируют полипептиды, которые связываются между собой, образуя фактор транскрипции CBF-la/CBF-1b, необходимый для нормального гемопоэза.
Перестройки t(8;21) и inv(16) приводят к образованию гибридных генов, кодирующих белки, препятствующие функционированию CBF-la/CBF-lb и блокирующие созревание миелоидных клеток. Однако следует отметить, что мыши с отсутствием гена CBF-1a или CBF-1b и мыши с «включенным» геном, экспрессирующим гибридный белок СВF-1а или СВF-1b, погибают от недостаточности гемопоэза, а не от лейкемии. Таким образом, генетические повреждения, просто блокирующие созревание миелоидных клеток-предшественников, сами по себе недостаточны для развития лейкемии.
Накапливается все больше данных о том, что мутантные тирозинкиназы могут взаимодействовать с аберрантными факторами транскрипции и вызывать ОМЛ. Одним из примеров может служить ОМЛ с t(15;17). Здесь транслокация t( 15; 17) создает другой гибридный ген PML-RAR-a, кодирующий белок RAR-а (рецептор ретиноевой кислоты а), соединенный с белком PML (именуемого по названию опухоли — промиелоцитарная лейкемия). В присутствии физиологического количества ретиноевой кислоты нормальный RAR-a взаимодействует с другими факторами транскрипции, активируя гены, необходимые для дифференцировки гранулоцитов. Однако вместо этого гибридный белок PML-RAR-a взаимодействует с репрессорами транскрипции, что приводит к ингибиции созревания гранулоцитов.
При ОМЛ с транслокацией t(15;17) часто происходят активирующие мутации FLT3 (рецептора тирозинкиназы, передающего сигналы, которые повышают пролиферацию и выживаемость опухолевых клеток). Комбинация белка PML-RAR-a и активированного FLT3 — мощный индуктор ОМЛ у мышей, хотя ни один из генов в отдельности не обладает таким свойством. Идентичные мутации гена FLT3 найдены и при других формах ОМЛ, особенно ассоциированных с мутациями гена NPM (нуклеофозмина). Активирующие мутации другого рецептора тирозинкиназы, c-KIT, обнаружены в « 25% случаев ОМЛ, ассоциированной с inv(16) или t(8;21). Таким образом, аберрантная активация тирозинкиназ является частым (а возможно, и универсальным) признаком ОМЛ.
Транслокация t(15;17) служит не только патогенным признаком, но также помогает выбрать необходимый метод лечения, т.к. опухоли с этой транслокацией реагируют на фармакологические дозы полностью-транс-ретиноевой кислоты (ATRA). ATRA связывается с гибридным белком PML-RAR-a и противодействует его ингибирующему влиянию на транскрипцию генов-мишеней. Примечательно, что активация транскрипции преодолевает блокаду дифференцировки, и в течение 1-2 сут неопластические промиелоциты начинают дифференцироваться в нейтрофилы, которые быстро гибнут.
Реакция на ATRA показывает, что основной эффект PML-RAR-a заключается в блокаде дифференцировки, и это пример одного из наиболее удачных методов таргетной терапии при злокачественных опухолях у человека.
е) Клинические признаки. Большинство пациентов обращается за помощью в течение нескольких недель или месяцев после появления симптомов, обусловленных анемией, нейтропенией и тромбоцитопенией. Наиболее типичными симптомами являются повышенная утомляемость, лихорадка и спонтанные кровотечения из слизистых оболочек и кожи. Напомним, что эти симптомы очень сходны с наблюдаемыми при ОЛЛ.
Тромбоцитопения приводит к появлению геморрагических заболеваний, нередко распространенных. Обычно наблюдаются петехии (точечные кровоизлияния в кожу) и экхимозы (слившиеся петехии), геморрагии в серозные оболочки, выстилающие полости тела и внутренние органы и кровоизлияния в слизистые оболочки десен и мочевого тракта. Прокоагулянты и фибринолитические факторы, высвобождаемые лейкозными клетками, особенно при ОМЛ с t(15;17), усиливают риск кровоточивости. Часто возникают инфекции, особенно полости рта, кожи, легких, почек, мочевого пузыря и толстой кишки, нередко появляются оппортунистические инфекции, вызываемые грибами, Pseudomonas spp. и микроорганизмами-комменсалами.
Признаки и симптомы, указывающие на вовлечение в процесс других, не костномозговых тканей, обычно менее выражены при ОМЛ по сравнению с ОЛЛ, однако опухоли с моноритарной дифференцировкой часто инфильтрируют кожу (гематодерматоз) и десны. Вероятно, это отражает нормальную тенденцию моноцитов к экстравазации в ткани. Центральная нервная система при ОМЛ поражается реже, чем при ОЛЛ. Иногда ОМЛ локализуется в мягких тканях, тогда ее называют по-разному: миелобластома, гранулоцитарная саркома или хлорома. Без системной терапии такие опухоли со временем неизбежно трансформируются в выраженную ОМЛ.
ж) Прогноз. Острая миелоидная лейкемия (ОМЛ) — трудно излечимое заболевание. В результате химиотерапии полная ремиссия наблюдается у 60% пациентов, однако только у 15-30% отсутствие симптомов сохраняется в течение 5 лет. ОМЛ с t(8;21) или inv(16) имеет относительно хороший прогноз при обычной химиотерапии, особенно в отсутствие мутаций c-KIT. И наоборот, прогноз неблагоприятен для ОМЛ, возникающей после МДС или генотоксической терапии, и для заболевания, манифестировавшего в пожилом возрасте, возможно, вследствие того, что в этих условиях болезнь развивается на фоне повреждений или потери КСК. Такие формы ОМЛ, а также рецидивирующую ОМЛ всех типов лечат с помощью трансплантации костного мозга, когда это возможно.
Можно надеяться, что новые методы, разработанные на основе более полного выяснения молекулярного патогенеза, изменят ситуацию к лучшему. Наиболее ярким примером является ОМЛ с t(15;17), которую лечат терапевтическими дозами ATRA в сочетании с обычной химиотерапией, а в последнее время — солями мышьяка, вызывающими деградацию PML-RAR-a. Разрабатываются новые терапевтические методы, направленные на другие молекулярные мишени при ОМЛ, в частности на активированные рецепторы тирозинкиназы FLT3 и c-KIT.
- Вернуться в оглавление раздела "Патофизиология"
Оглавление темы "Патогенез лимфом и лейкозов":- Механизмы развития (патогенез) волосатоклеточного лейкоза
- Механизмы развития (патогенез) неуточненной периферической Т-клеточной лимфомы
- Механизмы развития (патогенез) анапластической крупноклеточной лимфомы
- Механизмы развития (патогенез) Т-клеточной лейкемии-лимфомы взрослых
- Механизмы развития (патогенез) грибовидного микоза и синдрома Сезари
- Механизмы развития (патогенез) крупноклеточной гранулярной лимфоцитарной лейкемии
- Механизмы развития (патогенез) экстранодальной Т/NK клеточной лимфомы
- Механизмы развития (патогенез) лимфомы Ходжкина
- Классификация миелоидных неоплазий
- Механизмы развития (патогенез) острой миелоидной лейкемии