
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизм развития (патогенез) аномалий желчных протоков и кист печени
Описана гетерогенная группа патологических состояний, в основе которых лежит нарушение строения и уменьшение количества внутрипеченочных желчных протоков. Такие состояния могут быть выявлены случайно при лучевых методах исследования, во время оперативного вмешательства или аутопсии.
Первые симптомы этих патологических состояний проявляются в детском и подростковом возрасте и представлены гепатоспленомегалией и портальной гипертензией при отсутствии нарушений функции печени. К этой группе заболеваний относят:
(1) комплексы Мейен-бурга;
(2) поликистозную болезнь печени;
(3) врожденный фиброз печени;
(4) болезнь Кароли;
(5) синдром Алажиля.
а) Комплексы Мейенбурга представляют собой мелкие скопления в портальных трактах или рядом с ними умеренно расширенных желчных протоков, окруженных фиброзной, иногда гиалинизированной «муфтой». Эти скопления часто описывают как гамартомы желчных протоков. Комплексы Мейенбурга часто не имеют клинического значения, за исключением необходимости дифференцировать их с метастазами в печень.
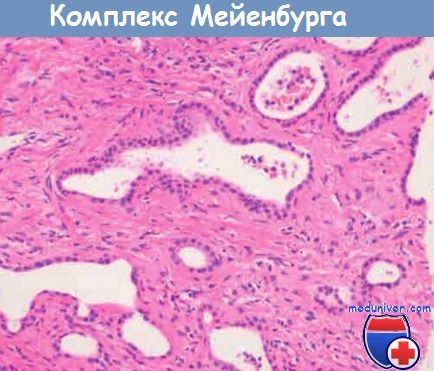
Отмечаются расширенные, неправильной формы желчные протоки.
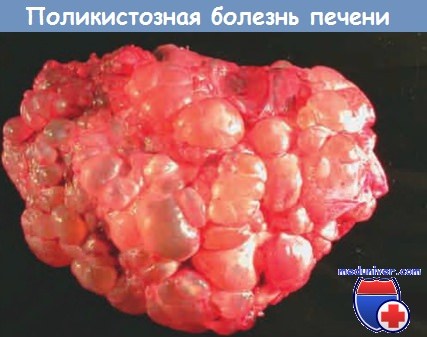
б) Поликистозная болезнь печени. При этом заболевании в печени выявляют множественные диффузно расположенные кисты; их число может варьировать от единиц до сотен. Кисты, выстланные кубическим или уплощенным эпителием, заполнены светло-желтым содержимым.
в) Врожденный фиброз печени. При этом состоянии портальные тракты увеличены за счет неровных широких пучков коллагеновых волокон, которые также образуют фиброзные септы, разделяющие ткань печени на неравномерные островки. В фиброзной ткани расположены желчные протоки неправильной формы, соединяющиеся с нормальными желчными протоками.
Такая аномалия развивается в связи с персистированием эмбриональных желчных протоков и последующим фиброзом портальных трактов, возникающим через определенный период времени. У лиц с врожденным фиброзом печени редко развивается цирроз, но могут возникать осложнения в виде портальной гипертензии и кровотечений из варикозно расширенных вен.
г) Болезнь Кароли. При этом заболевании крупные внутрипеченочные протоки сегментарно расширены и могут содержать густую желчь. Изолированные формы встречаются редко; обычно болезнь сочетается с фиброзом портальных трактов по типу врожденного фиброза печени. Заболевание часто осложняется внутрипеченочным холелитиазом, холангитом, абсцессами печени и портальной гипертензией. Лица с болезнью Кароли и врожденным фиброзом печени имеют повышенный риск развития холангиокарциномы.
Каждое из четырех описанных патологических: состояний может сочетаться с поликистозной болезнью почек. Наиболее частым экстраренальным проявлением аутосомно-доминантной поликистозной болезни почек, вызванной мутацией гена PKD1, являются одиночные или множественные кисты печени, развивающиеся у 75-90% пациентов с таким заболеванием почек. Поликистозная болезнь печени, вызванная мутацией гена PRKCSH (который кодирует основание 80К-Н протеинкиназы С), не сочетается с поликистозной болезнью почек.
Врожденный фиброз печени ассоциируется с аутосомно-рецессивной поликистозной болезнью почек, вызванной мутацией гена PKHD1 (поликистозная болезнь почек и печени). Точный патогенез этих заболеваний желчных путей и их связь с поликистозной болезнью почек остаются неясными.
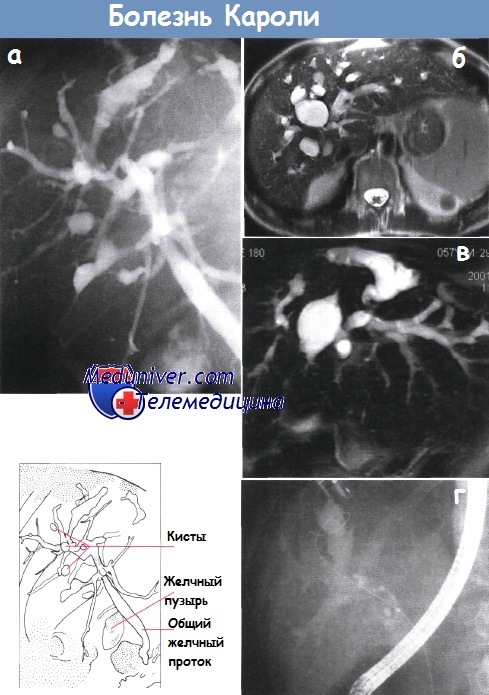
б - Множество расширенных внутрипеченочных желчных протоков. МРТ.
в - Магнитно-резонансная холангиография у того же пациента, что и на рис. а, подтвердила расширение желчных протоков.
г - Камень желчного протока, образовавшийся на фоне синдрома Кароли. Эндоскопическая ретроградная холангиопанкреатография.
д) Синдром Алажиля. Это редкое аутосомно-доми-нантное полиорганное заболевание, характеризующееся дисплазией внутрипеченочных желчных протоков (артериопеченочная дисплазия). Причина синдрома — мутации или делеции гена, расположенного на хромосоме 20р и кодирующего синтез белка Jagged1. Jagged1 является поверхностным клеточным белком, действующим как лиганд для рецептора Notch. Мутации Jagged выявляют у 94% лиц с клинически выявленным синдромом Алажиля, у остальных пациентов есть мутация рецептора Notch-2.
Сигнальный путь Jagged1/Notch регулирует метаболизм клетки и участвует в развитии систем органов, которые поражаются при синдроме Алажиля. Основные клинические признаки синдрома:
(1) хронический холестаз;
(2) периферический стеноз легочной артерии;
(3) бабочкообразные дефекты дужек позвонков;
(4) офтальмологический дефект, известный как задний эмбриотоксон;
(5) крупные черты лица у больного.
Пациенты могут доживать до зрелого возраста, но имеют высокий риск развития печеночной недостаточности и ГЦК.
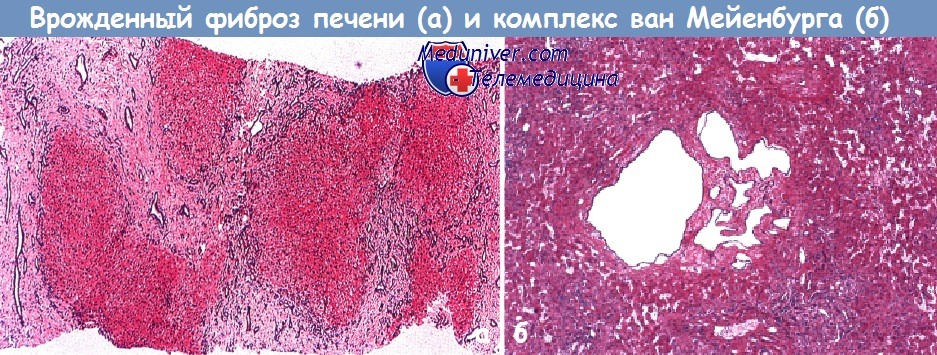
б - Комплекс ван Мейенбурга. Микроскопическая картина: небольшое скопление кистозно-расширенных мелких желчных протоков в портальных трактах. Протекает бессимптомно.
- Рекомендуем ознакомиться со следующей статьей "Механизм развития (патогенез) тромбоза воротной вены"
Оглавление темы "Патогенез болезней печени":- Механизм развития (патогенез) холестаза новорожденных
- Механизм развития (патогенез) вторичного билиарного цирроза печени
- Механизм развития (патогенез) первичного билиарного цирроза печени
- Механизм развития (патогенез) первичного склерозирующего холангита
- Механизм развития (патогенез) аномалий желчных протоков и кист печени
- Механизм развития (патогенез) тромбоза воротной вены
- Механизм развития (патогенез) нарушения внутрипеченочного кровотока
- Механизм развития (патогенез) синдрома Бадда-Киари - тромбоза печеночных вен
- Механизм развития (патогенез) синдрома обструкции синусоидов печени
- Механизм развития (патогенез) осложнений трансплантации печени