
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Миопатии - характеристика кратко
а) Мышечная дистрофия. Генетические МД характеризуются прогрессирующей дегенерацией мышц с замещением мышечных волокон соединительной тканью. Разные типы МД отличаются друг от друга клиническими проявлениями. Диагноз подтверждается биопсией мышцы или генетическим анализом. Прогноз зависит от типа МД.
Способов лечения МД не существует. Терапия может замедлить процесс и предупредить развитие контрактур и деформаций скелета, а также сохранить способность сидеть и ходить как можно дольше. Необходимо поддерживать нормальную массу тела. Лечебная физкультура направлена на предупреждение развития контрактур и респираторных нарушений. Пациенты с альвеолярной гиповентиляцией иногда нуждаются в неинвазивной вентиляции легких ночью. При болезни Дюшенна замедлить потерю мышечной силы могут кортикостероиды (преднизолон, дефлазакорт). Полезными могут быть ортезы (ночные шины, коленоголеностопные ортезы). При необходимости коррекции контрактур и сколиоза проводятся хирургические операции. При нарушениях сердечного ритма требуется имплантация водителя ритма.
Следует провести генетическую консультацию и проинформировать пациента о риске, связанном с общей анестезией. Общее и профессиональное обучение зависит от индивидуальных особенностей пациента.
1. МД Дюшенна. Заболевание начинается в раннем детстве. Постепенно развивается симметричная слабость проксимальных мышц (тазовый пояс, разгибатели голени → симптом Говерса) и их атрофия, которая распространяется дистально. Отмечаются задержка развития моторики, (псевдо-) гипертрофия икроножных мышц, кифосколиоз, гиперлордоз и слабость дыхательной мускулатуры. Примерно к 13 годам требуется инвалидное кресло. Результатом дистрофинопатии могут стать кардиомиопатия и когнитивные нарушения.
2. МД Беккера. Заболевание напоминает МД Дюшенна, но симптомы выражены слабее. Начало заболевания приходится на более поздний возраст, а способность ходить сохраняется дольше.
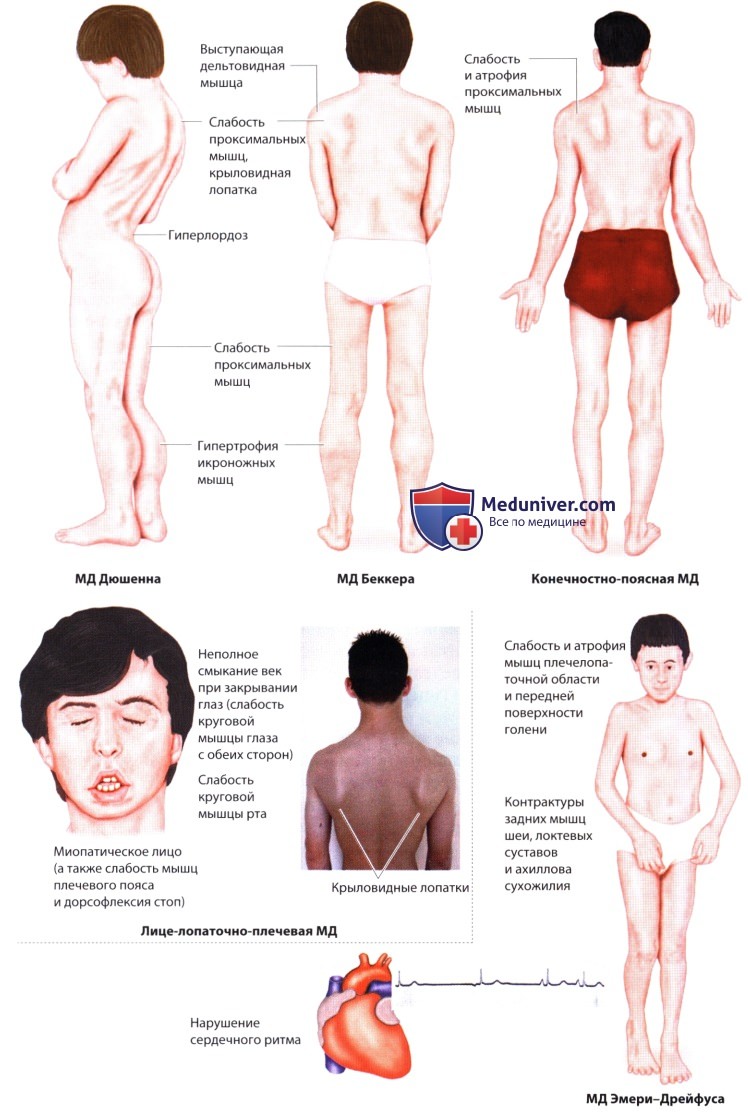
3. Конечностно-поясная МД. Слабость и атрофия развиваются в мышцах плечевого и/или тазового пояса. Существуют различные фенотипы, как с доминантным, так и с рецессивным наследованием. Для клинической классификации требуются биопсия мышцы и молекулярно-генетический анализ. Эти методы помогают отличить конечностно-поясную МД от других миопатий, в частности от дистрофинопатий, воспалительной миопатии и проксимальной миотонической миопатии.
4. Лице-лопаточно-плечевая МД. При этом типе асимметричной МД страдают мышцы лица и плеча (крыловидная лопатка), двуглавая и трехглавая мышцы плеча, мышцы тазового пояса и мышцы, осуществляющие дорсальную флексию стопы. Дельтовидная мышца обычно интактна.
5. МД Эмери-Дрейфуса характеризуется контрактурами суставов и слабостью плечеперонеальной группы мышц, которая позднее распространяется на область лопатки и тазовый пояс. Часто наблюдаются сердечно-сосудистые нарушения (непереносимость физических нагрузок, застойная сердечная недостаточность) с риском внезапной остановки сердца.
6. Миотоническая МД1 и МД2. См. отдельную статью на сайте - просим пользоваться формой поиска выше.
7. Окулофарингеальная МД. Начинается в пожилом возрасте. Возникает асимметричный птоз, частичная наружная офтальмоплегия и дисфагия.
б) Миотонические расстройства. Миотония характеризуется аномальной задержкой расслабления скелетных мышц (воспринимаемая как скованность) после их произвольного сокращения (миотония действия) или после перкуссии (перкуссионная миотония) с характерными ЭМГ-изменениями (нарастающе-убывающие или убывающие миотонические разряды). Особые формы миотонии различают по типу наследования, клиническим проявлениям и молекулярногенетическим признакам. При миотонии скованность уменьшается после повторных сокращений мышц (эффект разогрева). При парамиотонии скованность мышц, наоборот, возникает после физической нагрузки (парадоксальная миотония). Уровень креатин-киназы в сыворотке обычно в норме или немного повышен. Тяжелые миотонические реакции могут спровоцировать деполяризующие мышечные релаксанты и анестетики. Усилить миотонию могут также фенотерол, некоторые Р-блокаторы и диуретики.
Пациенты с легкой миотонией могут справляться с ее симптомами без лекарств. При тяжелой миотонии назначают мембраностабилизирующие препараты: флекаинид, мексилетин или карбамазепин. Проблемой могут стать побочные эффекты со стороны сердечно-сосудистой системы, особенно при миотонической дистрофии. Пациентам следует избегать переохлаждения.
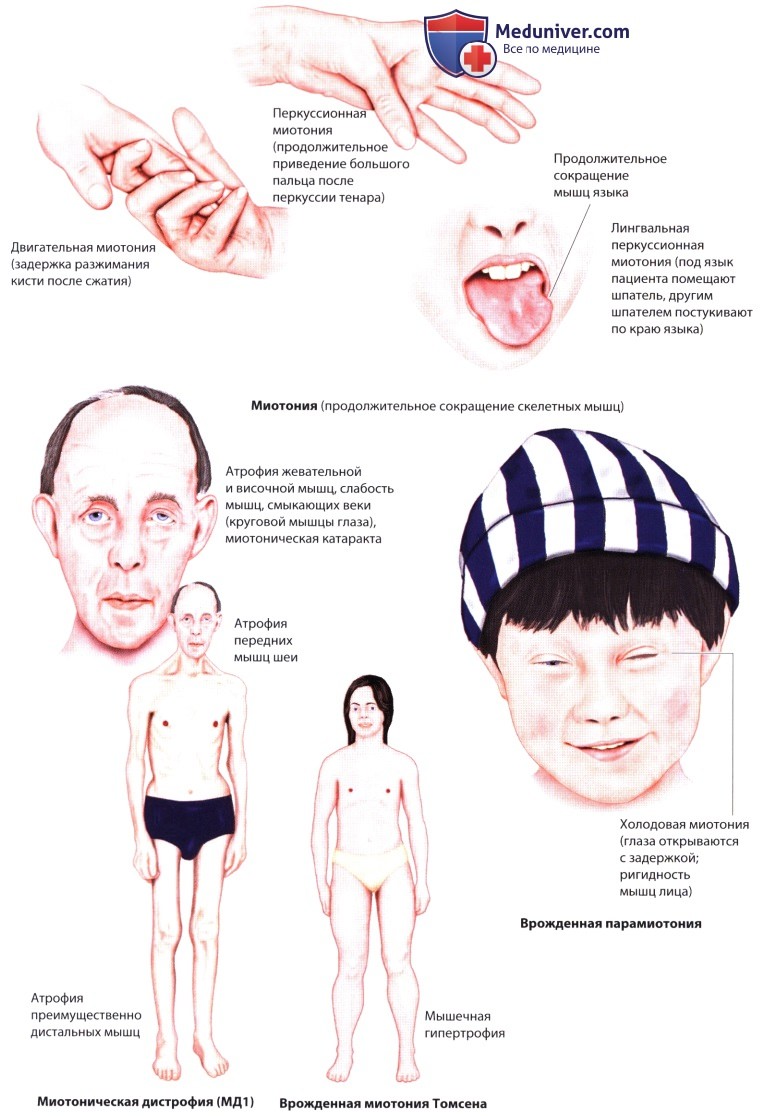
1. Недистрофические миотонические миопатии. В эту группу миотонических каналопатий входят врожденная доминантная миотония (тип Томсена), врожденная рецессивная миотония (тип Беккера), врожденная парамиотония (болезнь Эйленбурга), гиперкалиемический периодический паралич с миотонией и миотония, усиливающаяся приемом калия (миотония, спровоцированная приемом ацетазоламида). Эти миотонии обусловлены нарушениями натриевых или хлоридных каналов. При врожденной миотонии имеется мышечная гипертрофия. В случае врожденной парамиотонии за парадоксальной миотонией часто наступает слабость. Скованность усугубляется холодом. Парадоксальная миотония отчетливо наблюдается после повторного форсированного смыкания век.
2. Дистрофические миотонические миопатии. Миотоническая дистрофия взрослых 1-го типа (МД1), помимо прогрессирующей слабости, атрофии и миотонии дистальных мышц, характеризуется еще рядом симптомов. Это когнитивные нарушения, развитие катаракты в возрасте до 50 лет, сердечнососудистые проблемы (аритмии, блокады), апноэ во время сна, СД, новообразования и дисфагия. При МД2 (проксимальная миотоническая миопатия) мышцы не атрофируются, а слабость наблюдается преимущественно в проксимальных отделах конечностей наряду с миалгией. Имеются также внемышечные проявления.
в) Периодический паралич. При гипокалиемическом и гиперкалиемическом периодическом параличе (ПП), так же как при врожденной парамиотонии, возникают эпизоды вялого паралича различной продолжительности и тяжести. Бульбарная мускулатура, дыхательные и сердечная мышца не страдают. Гипокалиемический ПП обусловлен дисфункцией кальциевых каналов. При ЭМГ миотония между приступами не выявляется.
Если диагноз остается под вопросом, можно провести провокационные тесты. Развитие паралича после введения глюкозы с инсулином или только глюкозы указывает на гипокалиемический ПП; после введения калия или физической нагрузки - на гиперкалиемический ПП.
1. Лечение острых приступов. Легкие эпизоды мышечной слабости при гипокалиемическом ПП не требуют лечения. В более тяжелых случаях назначают калий перорально. Легкие эпизоды мышечной слабости при гиперкалиемическом ПП проходят в результате физических упражнений или введения углеводов. В более тяжелых случаях может потребоваться ингаляция сальбутамола или внутривенное введение глюконата кальция.
2. Профилактика. Гипокалиемический ПП: диета с низким содержанием соли и углеводов, интенсивные физические нагрузки противопоказаны, легкие - полезны. Пероральный прием ацетазоламида, спиронолактона или другого калийсберегающего диуретика. Гиперкалиемический ПП: диета с высоким содержанием углеводов, противопоказаны интенсивные физические нагрузки и холод. Перорально гидрохлоротиазид или ацетазоламид.
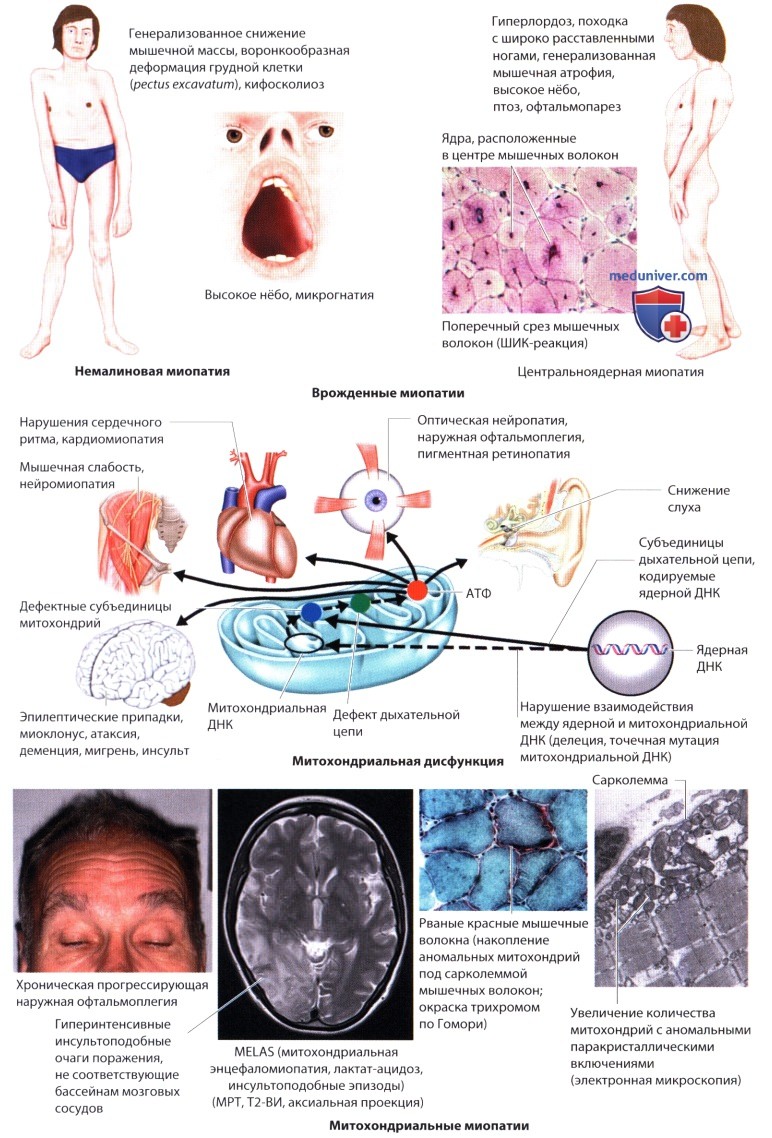
г) Врожденные миопатии. Отличительными признаками врожденной миопатии являются ее раннее начало (с рождения или в раннем детстве), симметричная генерализованная гипотония (вялый ребенок), часто дисморфия (высокое нёбо, дисплазия тазобедренного сустава, pes cavus - полая стопа, деформация грудной клетки) и характерные морфологические изменения мышц, которые обнаруживают при гистохимическом или электронно-микроскопическом исследовании биопсийного материала. Наблюдаются контрактуры, снижение или отсутствие рефлексов, вовлечение в процесс глазных, лицевых и дыхательных мышц. Развитие моторики происходит с задержкой. Миопатия медленно прогрессирует или остается стабильной. Некоторые миопатии диагностируются в дошкольном и раннем школьном возрасте или у подростков. Уровень креатинкиназы и данные ЭМГ почти не отличаются от нормальных показателей. Наследование может быть как доминантным, так и рецессивным либо Х-сцепленным. Классификация основана на генетических и характерных структурных аномалиях, выявляемых иммуногистохимическими методами. Например, при немалиновой миопатии, миопатии с накоплением миозина и миопатии с редуцированными тельцами, центральноядерной или миотубулярной миопатии обнаруживают накопление белков; при врожденной диспропорции волокон - мышечные волокна разного размера.
Гистологические изменения при врожденных МД сопоставимы с изменениями при дистрофической миопатии. В группу врожденных МД входят врожденная МД Фукуямы, врожденная МД Ульриха, болезнь «мышца-глаз-головной мозг», синдром Уокера-Варбург, врожденные интегрин-и мерозин-негативные МД. Врожденными бывают также миотоническая дистрофия МД1, лице-лопаточно-плечевая МД и МД Дюшенна.
д) Метаболические миопатии. Это гетерогенная группа заболеваний, вызванных наследственными аномалиями ферментов. «Динамические» миопатии характеризуются миалгией при физической нагрузке, слабостью, спазмами, миоглобинурией, а «статические» миопатии - постоянной или прогрессирующей мышечной слабостью.
Общим биохимическим признаком митохондриальных миопатий являются дисфункции дыхательной цепи митохондрий, β-окисления жирных кислот или нарушение обоих этих процессов.
Для синтеза АТФ в митохондриях наиболее важными субстратами служат пируват и жирные кислоты, которые подвергаются окислительному фосфорилированию ферментами дыхательной цепи (они расположены на внутренней мембране митохондрий). Р-окисление жирных кислот происходит в матриксе митохондрий. Структура ферментов дыхательной цепи кодируется как митохондриальной, так и ядерной ДНК. Наследование может быть материнским или аутосомным (мутация ядерной ДНК). Спорадические случаи возникают при мутациях митохондриальной ДНК.
При этих нарушениях могут страдать различные органы и системы: мышцы (сниженная выносливость, атрофия, спазмы, миоглобинурия), ЦНС (судороги, головная боль, поведенческие расстройства), зрение (птоз, наружная офтальмоплегия, пигментный ретинит), слух (нейросенсорная тугоухость), сердце (аритмия, сердечная недостаточность), ЖКТ (диарея, рвота), эндокринная система (СД, гипотиреоз), автономная нервная система (импотенция, повышенное потоотделение).
Диагноз ставится на основе клинических признаков, данных лабораторного исследования (повышение концентрации сывороточного лактата в покое; иногда концентрация лактата повышена и в ЦСЖ; его высокая концентрация длительно сохраняется после физической нагрузки), биопсии мышц (рваные красные мышечные волокна, иногда с дефицитом цитохром ооксидазы) и молекулярных исследований (анализ митохондриальной ДНК мышц, тромбоцитов, лейкоцитов). Способа лечения митохондриальных миопатий в настоящее время не существует.
е) Миастения (myasthenia gravis). Характерной особенностью МГ является мышечная слабость, которая возникает в результате физической нагрузки и обусловлена нарушением передачи нервного импульса в нервно-мышечных синапсах. Приблизительно у 80% пациентов в сыворотке обнаруживают антитела к н-холинорецепторам (никотиновым ацетилхолиновым рецепторам, АХР), а у 5-10% - антитела к специфической мышечной тирозинкиназе. У пациентов, в сыворотке которых отсутствуют названные антитела, недавно были обнаружены антитела к LRP4 (белку, родственному рецептору липопротеинов низкой плотности). При МГ важную роль играет тимус, вероятно, в результате генетического нарушения, которое препятствует стимуляции АХР. У 10-15% пациентов с МГ развивается тимома (па-ранеопластический синдром). Существуют также врожденные и семейные формы.
Клинические проявления. При миастении, вызванной поражением АХР, периодическая мышечная слабость впервые возникает в наружных мышцах глаза или проявляется птозом. В 15% случаев заболевание ограничивается этой областью (глазная форма миастении). У остальных пациентов заболевание в течение 2 лет распространяется на другие группы мышц и становится генерализованным (генерализованная миастения). Асимметричная мышечная слабость нарастает после физической нагрузки и уменьшается в покое. Могут поражаться мышцы лица и глотки (у 15% пациентов в начале заболевания). Результатом являются бедная мимика, дизартрия, затрудненные жевание, глотание и движения головы. Слабость дыхательных мышц и мышц глотки затрудняет откашливание и повышает риск аспирации. Постепенно пациенты утрачивают способность стоять или ходить. Миастению могут усиливать некоторые лекарственные препараты, инфекции, эмоциональный стресс, нарушение электролитного баланса, гормональные сдвиги и яркий свет (↑ диплопия, птоз). Часто миастении сопутствуют гипертиреоз, тиреоидит и аутоиммунные заболевания соединительной ткани. При миастении, связанной с наличием аутоантител к мышечной специфической тирозинкиназе, характерно поражение краниобульбарной мускулатуры: развивается слабость мышц лица, глотки, шеи и дыхательных мышц или генерализованная миастения. Миастенические или холинергические кризы могут угрожать жизни.
Диагностика. Диагноз подтверждается с помощью тестов, перечисленных в отдельной статье на сайте - просим пользоваться формой поиска выше.
Лечение. При глазной форме МГ назначают симптоматическое лечение ингибиторами ацетилхолинэстеразы (пиридостигмина бромид). При недостаточной эффективности добавляют кортикостероиды или азатиоприн. При генерализованной форме МГ назначают ингибиторы ацетилхолинэстеразы, кортикостероиды, азатиоприн или другие иммуносупрессоры (микофенолата мофетил, циклоспорин или циклофосфамид). Плазмаферез и внутривенное введение иммуноглобулинов назначаются при резком ухудшении состояния или при подготовке ослабленного пациента к тимэктомии. Тимэктомия обычно рекомендуется пациентам до 60 лет с умеренной или тяжелой мышечной слабостью и всем пациентам с тимомой, но не с глазной формой МГ. Дальнейшее лечение зависит от степени улучшения, достигнутого с помощью описанных мер. Большинство пациентов могут вести нормальный образ жизни, но нуждаются в пожизненной иммуносупрессии. Специальные мероприятия требуются во время миастенических или холинергических кризов, при тимоме, беременности, МГ новорожденных и врожденной МГ.
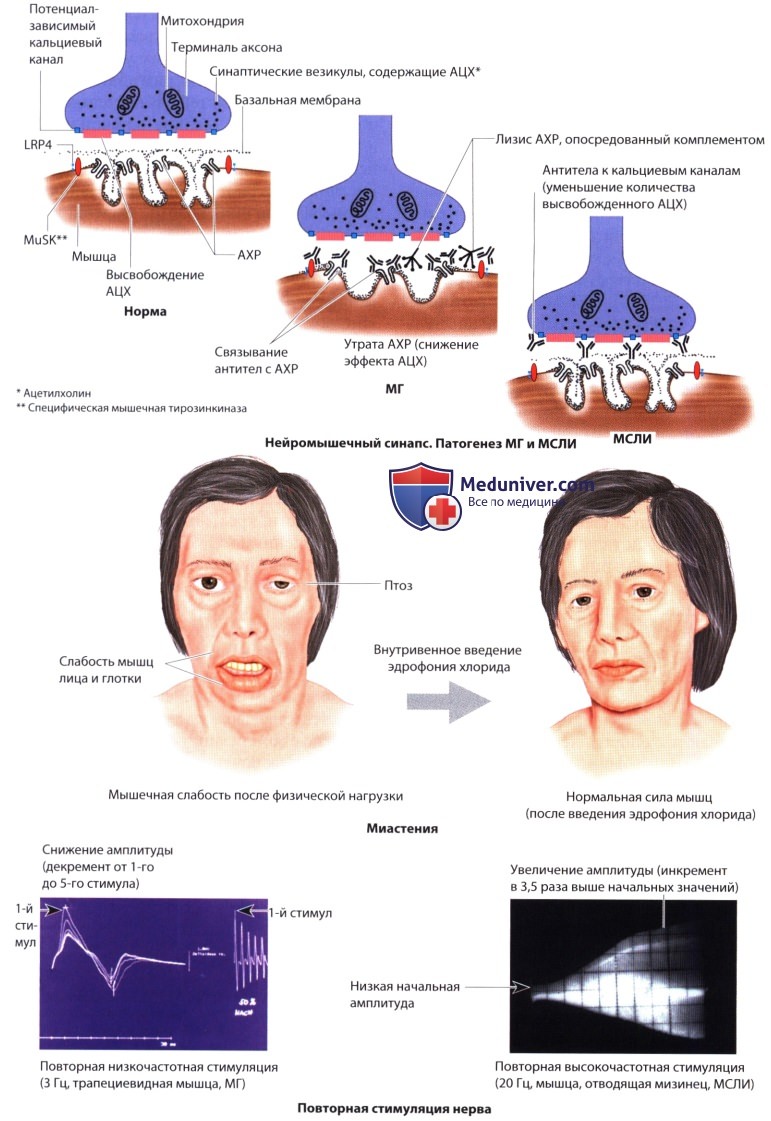
ж) Миастенический синдром Ламберта-Итона. Причиной миастенического синдрома Ламберта-Итона (МСЛИ) являются антитела к потенциалзависимым кальциевым каналам пресинаптического полюса нервно-мышечных синапсов и уменьшение высвобождения ацетилхолина. МСЛИ часто является паранеопластическим проявлением мелкоклеточного рака легкого и появляется раньше, чем обнаруживают опухоль. МСЛИ характеризуется слабостью проксимальных мышц (нижние конечности). Слабость уменьшается после кратковременной физической нагрузки, но усиливается при продолжительной. Отмечаются вегетативные симптомы (сухость во рту, постуральная гипотензия) и гипорефлексия. ЭМГ выявляет снижение амплитуды потенциалов действия некоторых мышц. Амплитуда увеличивается после кратковременного максимального произвольного сокращения или после повторной стимуляции с частотой 20-50 Гц.
Лечение: 3,4-диаминопиридин (увеличивает высвобождение ацетилхолина) и ингибиторы ацетилхолинэстеразы, плазмаферез, внутривенное введение иммуноглобулинов, терапия основного заболевания.
з) Воспалительные миопатии. В эту группу входят полимиозит, миозит с включениями телец и дерматомиозит.
Причина воспалительных миопатий неизвестна. Гистологически они характеризуются воспалением, фиброзом и утратой мышечных волокон.
Полимиозит и миозит с включениями телец, по-видимому, представляют собой процессы, опосредованные Т-лимфоцитами. Цитотоксические CD8+ Т-лимфоциты и макрофаги повреждают мышечные волокна, которые экспрессируют патогенные молекулы ГКГС-1. Комплексы CD8+/ГКГС-1 можно выявить специальными методами. Клеточные инфильтраты, по-видимому, вызывают деструкцию мышечных волокон. Депозиты амилоида при миозите с включением телец указывают на дегенеративный процесс.
Дерматомиозит, по-видимому, является гуморально опосредованным процессом. Первичными мишенями для антигенов служат компоненты сосудистого эндотелия. Повреждение эндотелия, деструкция капилляров, ишемия и микроинфаркты (→ периваскулярная атрофия) возникают после активации комплемента и мембранолитического комплекса С5b-9. Воспалительные инфильтраты, расположенные в эндомизии, содержат В-клетки, CD4+ Т-клетки, плазмоцитоидные дендритные клетки и макрофаги.
1. Полимиозит начинается постепенно со слабости проксимальных мышц нижних конечностей. По мере прогрессирования болезни слабость распространяется и на верхние конечности. Обычно страдают дельтовидные мышцы и мышцы-сгибатели шеи. Может развиться дисфагия. Пораженные мышцы в конечном счете атрофируются. Заболевают главным образом взрослые. Так как симптомы неспецифичны, требуется проведение обследования, чтобы отличить полимиозит от миозита с включениями телец, дистрофических и метаболических миопатий, заболевания моторных нейронов, заболеваний соединительной ткани, токсических миопатий, вирусных или бактериальных миозитов, эндокринопатий и некротизирующей аутоиммунной миопатии (связанной с применением статинов, злокачественными новообразованиями и вирусными инфекциями).
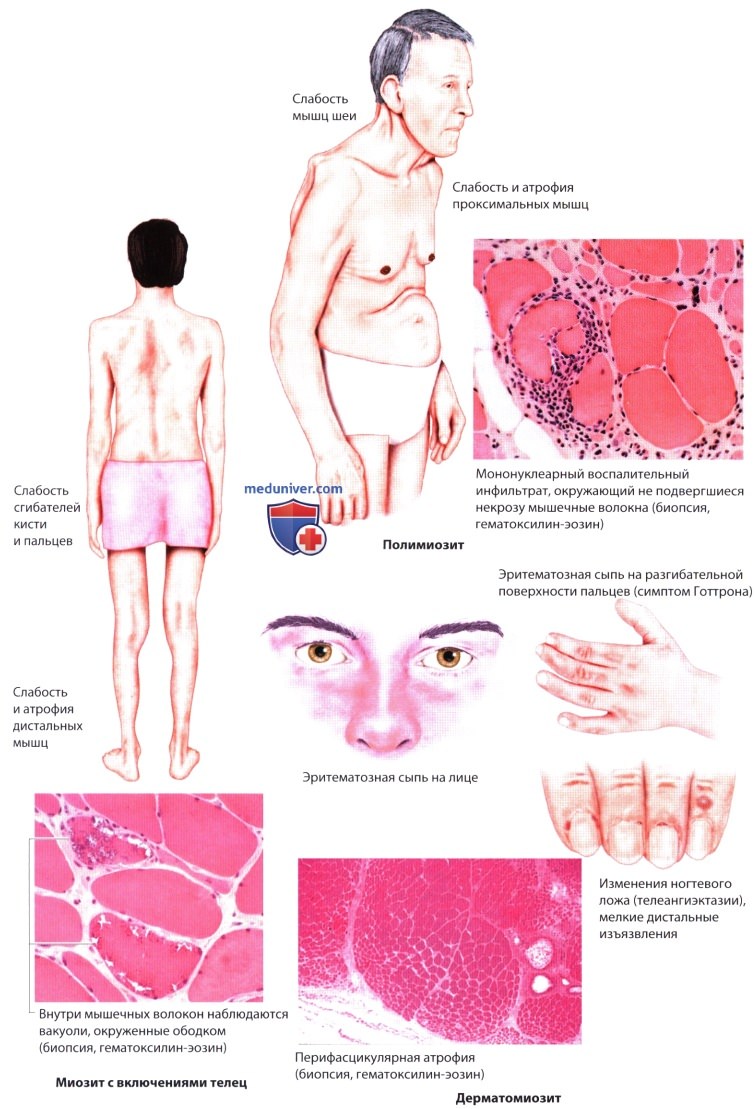
2. Миозит с включениями телец обычно начинается после 50 лет. Он характеризуется слабостью и атрофией (иногда асимметричной) дистальных мышц нижних конечностей (мышц, осуществляющих тыльное сгибание стопы, разгибание голени) и ранней утратой коленного рефлекса. Возможна слабость и атрофия мышц кисти, часто присутствует дисфагия. Дифференциальную диагностику проводят с полимиозитом, болезнью моторных нейронов и периферической нейропатией. Встречаются спорадические, наследственные и семейные формы.
3. Дерматомиозит встречается у детей и взрослых. Он прогрессирует быстрее, чем полимиозит, и отличается от последнего наличием голубовато-красной или фиолетовой сыпи на открытых участках кожи (веки, щеки, шея, грудь, разгибательные поверхности конечностей). В ногтевых ложах обнаруживаются мелкие кровоизлияния и телеангиэктазии. У детей могут возникать подкожные депозиты кальция. У 15% пациентов выявляются новообразования (рак яичника, молочной железы, кишечника, неходжкинская лимфома, меланома). Дерматомиозит также связан со склеродермией или сочетанием нескольких системных заболеваний соединительной ткани (перекрестный синдром - overlap-синдром). От дерматомиозита следует отличать эозинофильный фасциг (синдром Шульмана), характеризующийся болезненной индурацией кожи, отеком подкожной клетчатки и мышечной слабостью.
Диагноз миозита подтверждается обнаружением в сыворотке повышенной концентрации саркоплазматических ферментов (в частности, креатинкиназы), характерными изменениями ЭМГ и биопсией мышц. Участок для биопсии мышцы может быть выбран при помощи различных визуализирующих методов (КТ, МРТ, УЗИ).
Лечение. Полимиозит и дерматомиозит лечат с помощью иммуносупрессии, т.е. глюкокортикоидами, азатиоприном или внутривенным введением иммуноглобулинов. Лечебная физкультура начинается сразу после стабилизации состояния пациента. Миозит с включениями телец может отвечать на низкие дозы глюкокортикоидов или внутривенное введение иммуноглобулинов.
- Читать "Нервно-мышечные синдромы - характеристика кратко"
Редактор: Искандер Милевски. Дата публикации: 9.4.2020