
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Эффект Варбурга в метаболизме опухоли
Опухолевая клетка переключает свой метаболизм с эффективного митохондриального, с высоким потреблением кислорода, на гликолиз, невзирая на достаточные запасы кислорода.
Данный феномен, названный эффектом Варбурга и известный также как аэробный гликолиз, открыт много лет тому назад (за открытие данного эффекта Отто Варбург получил Нобелевскую премию в 1931 г.), но был забыт до недавнего времени. Такие метаболические изменения настолько типичны для опухолевых клеток, что рассматриваются некоторыми как восьмой маркер злокачественной опухоли.
В клинике способность опухоли выступать в роли «ловушки глюкозы» используют для визуализации методом позитронно-эмиссионной томографии. Для этого пациенту вводят 18F-фтордезоксиглюкозу (неметаболизируемое производное глюкозы), которая в основном захватывает опухолевые клетки (а также нормальные, активно делящиеся клетки костного мозга).
Позитронно-эмиссионная томография позволяет обнаружить большинство опухолей, особенно быстрорастущие. Тем не менее причинно-следственная связь между аэробным гликолизом и прогрессированием опухоли полностью не раскрыта, как и инициальное повреждение, вызывающее эти изменения.
Хорошо известно, что при аэробном гликолизе образуется 2 молекулы АТФ на 1 молекулу глюкозы, а при окислительном фосфорилировании в митохондриях генерируется более 20 молекул АТФ. Каким образом переход на менее эффективный гликолиз обеспечивает опухоли преимущества в росте? Для объяснения этого эффекта были предложены несколько взаимодополняющих гипотез. Одна из них объясняет эффект Варбурга как приспособление опухоли к выживанию в условиях гипоксии.
Несмотря на то что в опухоли идет усиленный ангиогенез, новообразованные сосуды сформированы неправильно, поэтому опухоль находится в состоянии более выраженной гипоксии по сравнению с нормальной тканью. Активация HIF-1a под действием гипоксии не только стимулирует ангиогенез, но и увеличивает экспрессию множества генов ферментов, участвующих в гликолизе, и снижает экспрессию генов окислительного фосфорилирования. Следовательно, происходит переход к режиму экономии.
Снижение потребности опухолевой клетки в кислороде приводит к относительному его увеличению, и питание получает большее количество опухолевых клеток, в результате рост опухоли продолжается.
Однако эффект Варбурга — это гликолиз, развивающийся при достаточном количестве кислорода для окислительного фосфорилирования. Таким образом, изменения, стимулирующие переключение метаболизма при гипоксии, должны быть зафиксированы в опухолевой клетке. Бывает, что период гипоксии сменяется нормоксией (что часто и происходит в опухолях), однако повышенный гликолиз в опухолевых клетках сохраняется.
Кроме того, мутации онкогенов и генов-супрессоров, стимулирующих рост, таких как RAS, р53, PTEN, также обусловливают метаболические изменения в клетках. Теперь рассмотрим, почему опухолевая клетка использует энергетически менее продуктивный источник снабжения.
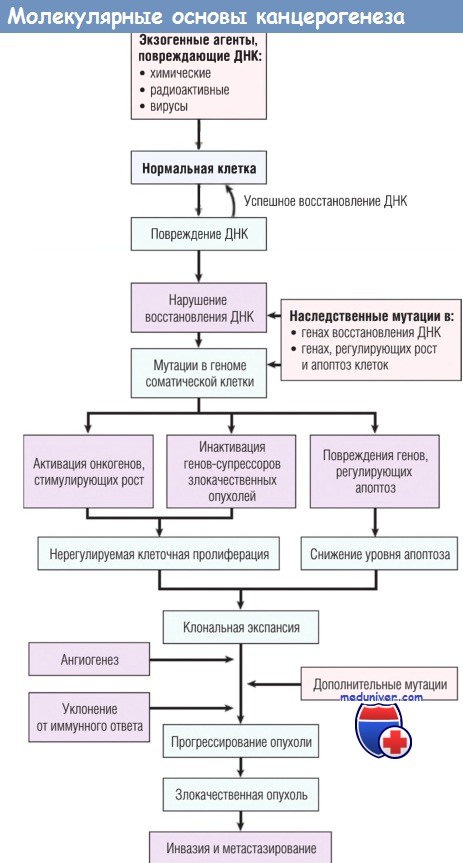
ДНК — дезоксирибонуклеиновая кислота.
В делящейся клетке (опухолевой и нормальной) удваиваются не только ДНК, но и другие компоненты, включая мембраны, белки и органеллы. Для этих процессов нужно повышенное количество питательных веществ, особенно глюкозы (которая обеспечивает энергией биосинтез этих компонентов) и аминокислот (используемых в качестве строительных блоков при синтезе белков).
Цикл окисления глюкозы до пирувата может быть переведен на анаболические пути, такие как синтез липидов и нуклеотидов, а опухолевые клетки могут перевести метаболизм глутамина на гликолиз и на анаболизм.
Таким образом, измененный метаболизм опухолевой клетки повышает ее возможности в синтезе строительных блоков, обеспечивающих деление и рост опухоли. Несомненно, что нарушения работы сигнальных путей онкогенеза, стимулируют захват глюкозы и других питательных веществ, большую активность гликолиза по сравнению с окислительным фосфорилированием и повышение анаболических реакций в опухолевой клетке.
В норме факторы роста стимулируют захват глюкозы и аминокислот через сигнальный путь PI3K/AKT/mTOR, начинающийся от тирозинкиназного рецептора и других рецепторов факторов роста; в опухолевых клетках эти сигналы автономны. Следовательно, мутации в онкогенах и генах-супрессорах активируют не только сигнальные пути, стимулирующие выживаемость и пролиферацию, но и процессы гликолиза и анаболизма с биосинтезом постоянных компонентов опухолевой клетки.
Сегодня на эффект Варбурга фактически посмотрели по-новому и обнаружили другие интереснейшие связи, свидетельствующие об участии онкопротеинов и генов-супрессоров. Примером служит участие гена-супрессора LKB1, кодирующего треонинкиназу, мутировавшего при синдроме Пейтца-Егерса, при котором развиваются доброкачественные и злокачественные опухоли ЖКТ.
Супрессивная активность гена осуществляется через активацию им АМФ-зависимой протеинкиназы — консервативного сенсора энергетического статуса клетки, являющегося важным негативным регулятором mTOR. Так, LKB1 подавляет образование опухоли, частично за счет нарушения анаболизма.
Достойны внимания два других гена-супрессора, мутирующие при туберозном склерозе, — TSC1 и TSC2, также являющиеся негативными регуляторами mTOR. С другой стороны, трансформирующий эффект многих онкопротеинов, включая мутантные тирозинкиназные рецепторы и хорошо известный онкогенный фактор транскрипции c-MYC, может стимулироваться отчасти индукцией эффекта Варбурга.
Такие взаимодействия обусловили множество попыток исследователей воздействовать на сигнальные пути, управляющие анаболизмом опухолевых клеток, в т.ч. на путь PI3K/AKT/mTOR.
Как обсуждалось ранее, в клетках существует множество барьеров для предотвращения нежелательного роста. Одним из адаптивных ответов нормальной клетки на дефицит кислорода и глюкозы является аутофагия — состояние, в котором клетка останавливает свой рост и осуществляет катаболизм собственных органелл, белков, а также мембран как источника углеводов для продукции энергии. Если адаптивная реакция недостаточна, то клетка погибает.
Часто создается впечатление, что опухолевые клетки могут расти в невероятных условиях без запуска аутофагии. Это наводит на мысль, что в опухолевых клетках сигнальные пути аутофагии нарушены. С этим согласуется тот факт, что часть генов, запускающих аутофагию, относятся к генам-супрессорам. Наиболее известный из них PTEN (негативный регулятор пути PI3K/AKT), мутирующий или «молчащий» в результате эпигенетических перестроек во многих опухолях.
Является ли аутофагия всегда злом для опухолевой клетки? Ответа на этот вопрос еще нет. Например, в условиях тяжелого дефицита питательных веществ опухолевые клетки могут использовать аутофагию для превращения в «спящие» клетки, характеризующиеся состоянием метаболической гибернации, что позволяет им выжить в течение длительного времени. Полагают, что именно такие клетки ответственны за неудачи в лечении злокачественных опухолей, т.к. становятся резистентными к методам терапии, воздействующим на активно делящиеся клетки.
Таким образом, аутофагия может быть «другом» или «врагом» опухолевых клеток в зависимости от того, как построены сигнальные пути в конкретной опухоли.
- Рекомендуем ознакомиться со следующей статьей "Хромосомные изменения при опухоли"
Оглавление темы "Патогенез опухоли":- Молекулярная генетика метастазирования опухоли
- Геномная трансформация как причина развития опухоли
- Влияние стромы на развитие опухоли
- Эффект Варбурга в метаболизме опухоли
- Хромосомные изменения при опухоли
- Амплификация генов при опухоли
- Эпигенетические изменения как причина опухоли
- МикроРНК как причина злокачественной опухоли
- Молекулярные механизмы многоступенчатого канцерогенеза
- Механизмы и фазы химического канцерогенеза