
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Злокачественный атрофический папулез (болезнь Дегоса): причины, клиника, диагностика, лечение
Злокачественный атрофический папулез - краткий обзор:
• Редкое заболевание, преимущественно носящее вазоокклюзирующий характер, поражающее в первую очередь кожу, желудочно-кишечный тракт и центральную нервную систему.
• Диагноз выставляется на основе клинико-патологических данных.
• Характеризуется появлением многочисленных типичных фарфорно-белых атрофических папул, окруженных ободком розовой эритемы и/или телеангиэктазиями.
• При патологоанатомическом исследовании выявляются участки некроза дермы клиновидной формы с отеком и отложением муцина.
• Существуют доброкачественные формы без внекожного поражения.
В начале 40-х годов XX века злокачественный папулез впервые описал Кельмейер (Kohlnteier), а год спустя Дегос, Делор и Трико выделили это заболевание как отдельную нозологическую единицу.1'3 В настоящее время известно, что характерные клинические изменения болезни Дегоса являются признаком кожной тромбо-облигерирующей васкулопатии, а не отдельным заболеванием. В действительности, такие изменения могут обнаруживаться по меньшей мере при двух различных клинических состояниях: (1) при идиопатических заболеваниях (как классической болезни Дегоса, так и ее доброкачественном варианте) и (2) в качестве дополнительных проявлений при различных болезнях соединительной ткани, таких как антифосфолипидный синдром, системная красная волчанка, дерматомиозит и системный склероз.
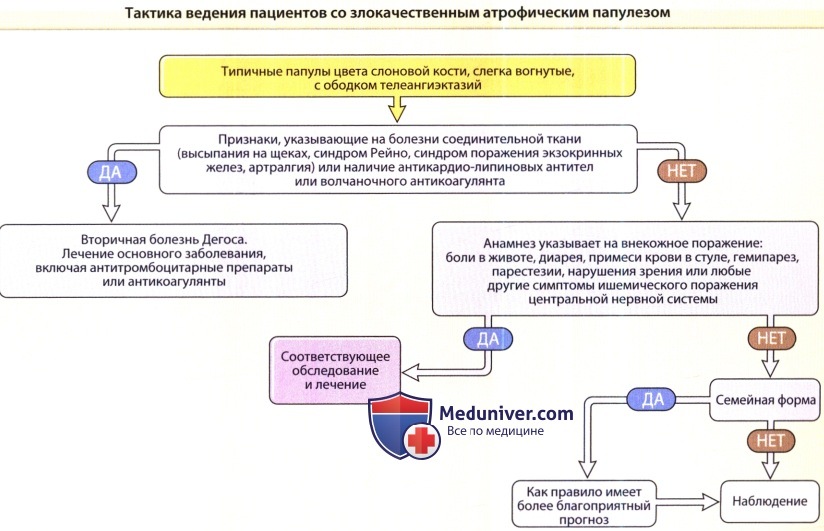
а) Эпидемиология злокачественного атрофического папулеза. Классическая болезнь Дегоса встречается очень редко, описано менее 200 случаев. Почти всегда она поражает лиц европеоидной расы, однако заболевание было выявлено и у афроамериканцев, и среди жителей Японии. Чаще всего заболевание проявляется в течение третьего и четвертого десятилетий жизни, однако может развиваться в любом возрасте. Мужчины болеют чаще, чем женщины (в соотношении 3:1). Большинство случаев — спорадические, однако были описаны и семейные варианты. Большинство семейных случаев связаны с аутосомно-доминантным типом наследования.
б) Этиология и патогенез злокачественного атрофического папулеза. Этиология болезни Дегоса неизвестна. Гистологические изменения, выявляемые у пациентов со злокачественным атрофическим папулезом, позволяют предположить преимущественное участие в патологическом процессе окклюзии сосудов. Таким образом, в качестве основного патогенетического механизма должны рассматриваться васкулярная коагулопатия и/или повреждение эндотелиальных клеток. В прогрессировании болезни, возможно, играет роль комбинация протромботических факторов. У некоторых пациентов с болезнью Дегоса проводились обширные исследования протромботических факторов, однако ни одного повторяющегося нарушения выявлено не было. Рекомендован скрининг антифосфолипидных антител/волчаночного коагулянта и криоглобулинов, однако Assier и соавт. не выявили наличия этих маркеров в своих работах, включающих 15 пациентов. У некоторых пациентов было выявлено ингибирование фибринолиза и тромбоцитарные нарушения, включая повышенную адгезию тромбоцитов и спонтанную агрегацию.
Симптомы болезни Дегоса следует рассматривать как кожные проявления, внутрисосудистого тромбоза. Несколько патологических процессов могут объединяться с формированием таких клинических и гистологических признаков.
в) Клиническая картина злокачественного атрофического папулеза:
1. Анамнез. Пациенты, как правило, обращают внимание на появление небольших изменений кожи, которые обычно не сопровождаются зудом или болезненностью. В некоторых случаях при сборе анамнеза и/или обследовании органов и систем будут выявлены признаки, указывающие на внекожную ишемию: боли в животе, диарея, мелена, тошнота, нарушение зрения, гемипарез, парестезии. Данные анамнеза также могут навести на мысль о возможности предшествующих случаев тромбоэмболии, которые позволят предположить наличие анти-фосфолипидного синдрома.
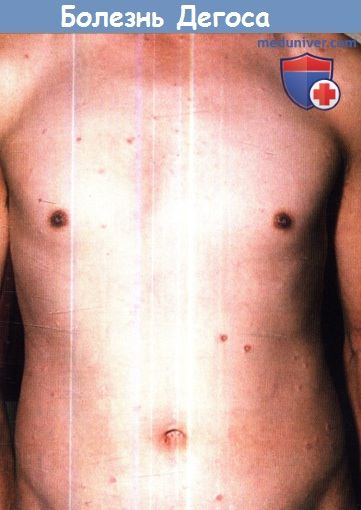
2. Кожные проявления. Изменения кожи начинают проявляться в виде розовых или цвета нормальной кожи пятнистых высыпаний размером 2-10 мм, преимущественно бессимптомных или сопровождающихся умеренным зудом, которые вскоре преобразуются в округлые, гладкие, часто куполообразной формы плотные папулы. У некоторых элементов может отмечаться центральное пупковидное вдавление и/или некроз. С течением дней или недель эти высыпания развиваются до фарфорно-белых атрофических папул с ободком розовой эритемы и/или телеангиэктазиями. Таким образом, элементы в стадии разгара сильно напоминают поражения, характерные для белой атрофии кожи (atrophie blanche).
Со временем красноватый контур исчезает, оставляя только белые рубцы, схожие со следами перенесенной ветряной оспы. Обычно патологические элементы отделены друг от друга, однако они могут сливаться с образованием полициклических участков атрофии или кожных изъязвлений.
Патологические элементы локализуются на коже туловища и конечностей. Ладони, ступни, лицо, волосистая часть кожи головы и наружные половые органы обычно не поражаются, хотя возможны исключения из этого правила. Сообщалось о линейном распространении поражений. Возможно вовлечение в патологический процесс глаз, при этом наиболее частым проявлением является образование аваскулярных участков на конъюнктиве, но поражаться могут также склеры, эписклера, сетчатка, сосудистая оболочка и зрительные нервы.
3. Классическая болезнь Дегоса. При классической болезни Дегоса число кожных элементов варьирует от нескольких единиц до одной сотни и более. Кожные изменения обычно предшествуют системным симптомам, которые могут отражать поражение желудочно-кишечного тракта (в том числе с перфорацией кишечника и развитием перитонита) и/или центральной нервной системы (с развитием геморрагического или ишемического инсульта). В редких случаях кожные изменения появляются одновременно или после симптомов поражения желудочно-кишечного тракта или центральной нервной системы.
Патологоанатомические исследования при аутопсии позволили выявить тромботические поражения мелких сосудов многочисленных органов, включая почки, мочевой пузырь, предстательную железу, печень, плевру, перикард, легкие и глаза.
У некоторых пациентов с классической болезнью Дегоса выявлялись антифосфолипидные антитела, что указывает на возможную связь данного заболевания с первичным антифосфолипидным синдромом.
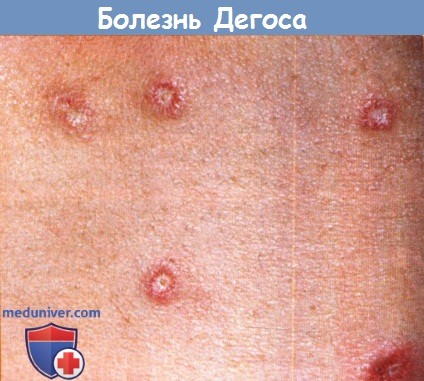

4. Доброкачественная болезнь Дегоса. Доброкачественная форма болезни Дегоса широко признана в настоящее время. При данном состоянии выявляется только кожное поражение. Большинство семейных случаев являются доброкачественными. У пациентов с антифосфолипидными антителами было описано развитие доброкачественной болезни Дегоса во время беременности, кроме того, характерные кожные изменения были описаны у пациента с синдромом приобретенного иммунодефицита. Авторам известен случай развития болезни Дегоса после инъекций интерферона, который может провоцировать развитие поражений наподобие белой атрофии кожи и микроангиопатии (L. Thomas, личная информация).
Четких различий между классической и доброкачественной формами болезни Дегоса не существует, и невозможно предсказать, у каких пациентов разовьется поражение внутренних органов, а у каких нет.
5. Патологические изменения по типу болезни Дегоса или вторичная болезнь Дегоса. Патологические изменения по типу болезни Дегоса могут встречаться у пациентов с установленной красной волчанкой, в особенности у лиц с положительными антифосфолипидными антителами, дерматомиозитом и системным склерозом.
г) Анализы при злокачественном атрофическом папулезе (болезни Дегоса):
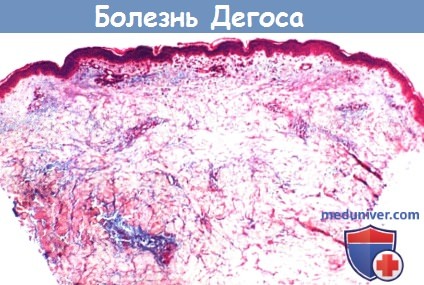
1. Гистологическая картина. Патологические элементы в стадии разгара представляют собой бедные клетками клиновидные участки некроза различной степени выраженности в сочетании с отеком дермы и обширными отложениями муцина, распространяющиеся от сосочкового до сетчатого слоя дермы. В основании пораженного очага могут выявляться окклюзированные сосуды с единичными участками некроза, утолщением сосудистых стенок и редкими участками периваскулярных лимфоцитарных инфильтратов. Однако картина полного лейкоцитарного васкулита никогда не выявляется у пациентов со злокачественным атрофическим папулезом.
Таким образом, данное заболевание не следует относить к васкулитам. Могут выявляться множественные вторичные изменения эпидермиса: гиперкератоз, поверхностный дерматит, разрозненные некротические кератиноциты и атрофия. У одного пациента был описан сочетанный склерозирующий панникулит. Результаты прямых иммунофлуоресцентных анализов оказались неоднозначными. При электронной микроскопии в некоторых случаях были выявлены тубулоретикулярные скопления внутри эндотелиальных клеток, значение которых неясно.
Так как некоторые гистопатологические признаки, такие как инфильтрация дермы муцином, поверхностный дерматит и периваскулярные лимфоцитарные инфильтраты в дерме, являются общими для системной красной волчанки и дерматомиозита, некоторыми авторами болезнь Дегоса рассматривается как возможный вариант красной волчанки.
2. Другие лабораторные и методы исследования. Специфических диагностических или прогностических маркеров болезни не существует. Исследование на наличие антиядерных антител, волчаночного антикоагулянта и антикардиолипиновых антител следует проводить каждому пациенту. Иногда можно выявить нарушения при расширенном исследовании гемостаза. При внезапном начале заболевания следует подозревать инфекцию парвовируса В19 и искать методом ПЦР вирусную циркулирующую ДНК.
Клиническое обследование может подтвердить диагноз вторичной болезни Дегоса при наличии признаков, указывающих на болезнь соединительной ткани или антифосфолипидного синдрома. Если при клиническом обследовании выявляются признаки поражения кишечника или головного мозга (боль, диарея, гемипарез, нарушения зрения, парестезия), необходимо проведение дополнительных методов исследования, (магнитно-резонансная томография головного мозга и/или эндоскопия/лапароскония).
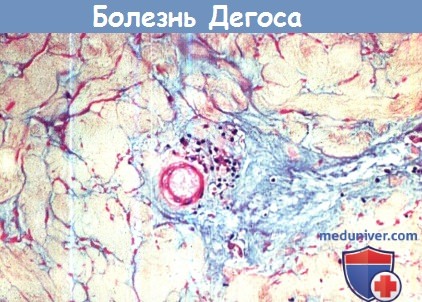
д) Диагностика. Диагноз обычно устанавливается на основании клинических данных, так как патологические элементы являются весьма характерными. С целью подтверждения диагноза необходима биопсия.
е) Осложнения. Основными осложнениями болезни Дегоса являются ишемические поражения головного мозга или желудочно-кишечного тракта, ведущие к летальному исходу. Наиболее частыми и неблагоприятными осложнениями являются желудочно-кишечные кровотечения, перфорации и перитонит. Реже встречаются неврологические осложнения, такие как гемипарез, афазия, поражение черепных нервов, моноплегия, сенсорные нарушения и судороги. В некоторых случаях прогрессирующее неврологическое поражение может привести к летальному исходу, особенно у детей. В редких случаях причиной летального исхода являются дыхательная недостаточность или инфаркт миокарда.
ж) Прогноз и течение. Несмотря на то что отсутствие признаков вовлечения внутренних органов в патологический процесс в течение двух лет после установления диагноза предвещает более благоприятный прогноз, системное поражение может развиваться спустя несколько лет после появления первых патологических элементов на коже и, таким образом, предсказать исход заболевания невозможно. Летальность при наличии внекожных проявлений превышает 50%, большинство таких пациентов погибает в течение 2-3 лет преимущественно в связи с тяжелым поражением кишечника. При семейном варианте болезни Дегоса прогноз более благоприятный.


з) Лечение. В связи с редкой встречаемостью болезни Дегоса не было проведено ни одного контролируемого клинического исследования, об этом заболевании получены лишь единичные сообщения. И все же, так как болезнь Дегоса в некотором смысле напоминает ливедоидную васкулопатию, возможно применение некоторых методов лечения, эффективных у пациентов с подобными состояниями.
Терапия при вторичной болезни Дегоса у пациентов с заболеваниями соединительной ткани должна, по нашему мнению, в сочетании со стандартными методами, включать антитромбоцитарные препараты.
Пациентам с классической болезнью Дегоса следует проводить обследование, направленное на выявление всех известных сердечно-сосудистых факторов риска. Больные должны прекратить курение. Кроме того, необходимо контролировать уровень липидов и при необходимости применять статины.
Терапия первой линии пациентов с болезнью Дегоса должна включать ингибиторы агрегации тромбоцитов (аспирин, клопидогрель,дипиридамол). Значительное число пациентов отвечают на терапию аспирином и дипиридамолом. С различными результатами в единичных случаях применялись антибактериальные препараты, хлорохин, иммуносупрессанты, фенилбутазон и фибринолитики. Было показано, что системные стероиды усугубляют течение болезни и не должны применяться. Различной эффективностью обладают гепарин и другие антикоагулянтные препараты, такие как антивитамины К (варфарин, флуиндион).
Однако по аналогии с ливедоидной васкулопатией, при острой болезни Дегоса для случаев, не отвечающих на терапию антитромбоцитарными средствами, могут потребоваться исследования эффективности низкомолекулярного гепарина, а также новых антикоагулятных препаратов. Внутривенные иммуноглобулины рекомендуются пациентам, не отвечающим на другие методы терапии или же в случае острого заболевания.
и) Список литературы:
- Kohlmeier W: Multiple hautnekrosen bei thrombangiitis obliterans. Arch Dermatol Syphil 181:783, 1941
- Degos R, Delort J, Tricot R: Papulose atrophiante maligne (syndrome cutaneo-intestinal mortel). Bull Mem SocMed Нфр Paris 64:803, 1948
- Pinault AL et al: Forme familiale bftnigne de maladie de Degos. Ann Dermatol Venereol 131:989, 2004
- Caux F et al: Anomalies de la fibrinolyse dans la maladie de Degos. Ann Dermatol Venerol 121:537, 1994
- Drucker CR: Malignant atrophic papulosis: Response to antiplatelet therapy. Dermatologica 180:90, 1990
- Torrelo A et al: Malignant atrophic papulosis in an infant. Br J Dermatol 146:916, 2002
- Kirkup ME et al: Inflammatory linear vasculopathy mimicking Degos disease. Br J Dermatol 150:1212, 2004
- Howsden SM et al: Malignant atrophic papulosis of Degos. Arch Dermatol 112:1582, 1976
- Bogenrieder T et al: Benign Degos’ disease developing during pregnancy and followed for 10 years. Acta Dermb Venereol 82:284, 2002
- Requena L, Farina C, Barat A: Degos disease in a patient with acquired immunodeficiency syndrome. J Am AcadDermatol 38:852, 1998
- Bugatti L et al: Atrophie blanche associated with interferon- alfa adjuvant therapy for melanoma: A cutaneous side effect related to the procoagulant activity of interferon? Dermatology 204:154, 2002
- Zuber J et al: Alpha-interferon-associated thrombotic microangiopathy. A clinicopathologic study of 8 patients and review of the literature. Medicine 81:321, 2002
- Stephansson EA et al: Lupus anticoagulant and the skin. A longterm follow-up study of SLE patients with special reference to histopathological findings. Acta Derm Venereol 71:416, 1991
- Dyrsen ME et al: Parvovirus B19-associated catastrophic endotheliatis with a Degos-like presentation. J Cutan Pathol 35:20-25, 2008
- Grilli R et al: Panniculitis mimicking lupus erythematosus profundus: A new histopathologic finding in malignant atrophic papulosis (Degos disease). Am J Dermatopathol 21:365, 1999
- Burg G et al: Maligne atrophische papulose (Morbus Khlmeier-Degos). Hautarzt 40:480, 1989
- Hairston BR et al: Treatment of livedoid vasculopathy with low-molecular-weight heparin: Report of 2 cases. Arch Dermatol 139:987, 2003
- Рекомендуем далее ознакомиться со статьей "Классификация сосудистых мальформаций кожи"
Редактор: Искандер Милевски. Дата публикации: 4.3.2019