
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Симптомы мышечной дистрофии и ее лечение
Мышечные дистрофии — группа, включающая около 30 редких наследственных заболеваний, характеризующаяся мышечной слабостью и дистрофией. Патологические изменения заключаются в мальформации мышечных волокон, гибели миоцитов с последующим их замещением соединительной и жировой тканью.
Данная группа заболеваний классифицируется в соответствии с формой наследования, возрастом начала заболевания, анатомической областью поврежденной мускулатуры, выраженностью мышечной слабости. Наиболее частыми формами заболеваний, встречающимися в ортопедической практике, являются:
• Мышечная дистрофия Дюшена — тяжелое, генерализованное, сцепленное с полом заболевание, возникающее у мальчиков в раннем детстве. Мышечная дистрофия Беккера — похожее, но менее тяжелое заболевание, дебютирующее позднее и намного медленнее прогрессирующее.
• Конечностно-поясничные мышечные дистрофии представляют смешанную группу заболеваний, обычно с аутосомно-рецессивным наследованием, с более локализованными изменениями, поражая детей (как мальчиков, так и девочек) в старшем возрасте.
• Лице-лопаточно-плечевая дистрофия — аутосомно-доминантное состояние различной степени тяжести, проявляющееся обычно в раннем детстве.

а) Мышечная дистрофия Дюшена. Прогрессирующее, сцепленное с полом, рецессивно наследуемое заболевание. Встречается с частотой 1 на 3500 родившихся мальчиков (или девочек с хромосомными нарушениями). Некоторые женщины являются «носителями заболевания с проявлениями», демонстрируя легкую мышечную слабость и судороги.
Дефект при данном заболевании определяется в локусе р21 Х-хромосомы и повреждает код гена белка дистрофина, отвечающего за поддержание целостности кардиомиоцитов клеток и поперечно-полосатой мускулатуры. Отсутствие активного дистрофина приводит к повышению проницаемости клеточной мембраны миоцитов, их повреждению и замещению жировой и соединительной тканью.
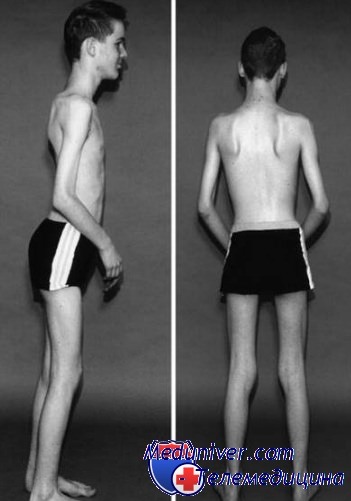
Клиническая картина. Клинические проявления становятся очевидными, когда ребенок начинает ходить. Он с трудом стоит и взбирается по лестнице, не способен правильно бегать и часто падает. Слабость начинается в мышцах проксимального отдела нижних конечностей и распространяется в дистальном направлении, поражая ягодичные, четырехглавые, передние большеберцовые мышцы, что приводит к широкой стойке и походке, при которой стопы находятся в эквинусном положении, таз наклонен вперед, спина изогнута в лордозе, шея в положении разгибания.
Икроножные мышцы выглядят объемными, но в большей степени из-за замещения жировой тканью и псевдогипертрофии, которая резко контрастирует с их явной слабостью. Характерный симптом заболевания—ребенок встает с пола, «взбираясь» вверх по собственным ногам (симптом Говерса); это связано со слабостью больших ягодичных мышц и мышц бедра.
Слабость мышц пояса верхних конечностей проявляется примерно через пять лет после дебюта заболевания, осложняя ходьбу с костылями. Мимические мышцы поражаются позже. К 10-летнему возрасту ребенок обычно теряет способность к самостоятельной ходьбе и становится зависим от инвалидного кресла, что в свою очередь провоцирует развитие сколиоза и впоследствии ухудшение дыхательной функции. Обычно причиной смерти является сердечно-легочная недостаточность, как правило, в возрасте до 30 лет.
Диагностика. Диагноз ставится на основании клинической картины, данных семейного анамнеза и исследовании концентрации креатининфосфокиназы в сыворотке, превышающей норму в 200-300 раз на ранних стадиях заболевания (у женщин-носителей уровень также повышен, но в меньшей степени). Диагноз подтверждается по результатам биопсии мышц и генетического тестирования с ДНК полимеразной цепной реакции.
Лечение. До тех пор, пока ребенок сохраняет способность ходить, физиотерапия, шинирование или операции на сухожилиях могут предотвратить или исправить деформации суставов и, таким образом, продлить период двигательной активности.
Применение кортикостероидов позволяет сохранить мышечную силу, но существуют выраженные побочные эффекты: остеопороз, повышенный риск переломов, формирование катаракты.
Экспериментальные исследования доказали эффективность использования дистрофина в форме миобластов, вводимых в пораженную мышцу, на лабораторных животных, но не на людях. Были также попытки применения генной терапии, но возникали трудности с вирусными векторами и связанными с ними иммунологическими ответами.
При выраженной сколиотической деформации (более 30°) эндокорректоры и спондилодез помогают поддерживать дыхательную функцию и улучшить качество жизни, но не всегда ее продолжительность. На доонерационном этапе должна оцениваться функция сердечно-сосудистой и дыхательной систем.
Важны семейные консультации. У 20% семей к моменту постановки диагноза пробанду уже есть младший сиблинг с еще неразвившимся заболеванием.
б) Мышечная дистрофия Бекера. Рецессивно наследуемое, также сцепленное с Х-хромосомой заболевание сходно с дистрофией Дюшена, но протекает легче. Уровень дистрофина снижен и/или носит аномальный характер. Больные (мужского пола) сохраняют способность ходить до подросткового возраста и доживают до среднего возраста. Мимические мышцы не поражаются, функция мускулатуры кишечника и мочевого пузыря, а также глотательная функция сохраняются.
в) Дистрофия пояса конечностей. Эта форма мышечной дистрофии, характеризующаяся слабостью пояса верхних и нижних конечностей, представляет гетерогенную группу состояний, большинство из которых наследуется аутосомно-рецессивно и не связано с полом.
Симптомы появляются в позднем подростковом возрасте. Слабость мышц пояса нижних конечностей вызывает ковыляющую походку и трудности при подъеме с низкого стула; при несостоятельности мышц пояса верхних конечностей затруднительно поднимать руки над головой. Однако мимические мышцы сохраняют свою функцию. Прогрессирование заболевание обычно медленное. (NB: вышеперечисленные симптомы могут быть ошибочно приняты за легкую форму спинальной мышечной атрофии).
Лечение заключается в физиотерапии и шинировании для предотвращения контрактур, а также в оперативном лечении. Так как дельтовидные мышцы сохранены, движения верхних конечностях возможны при фиксации лопатки к ребрам по задней поверхности грудной клетки для улучшения рычага дельтовидной мышцы.
г) Лице-лопаточно-плечевая дистрофия. Аутосомно-доминантное состояние с разнообразными проявлениями. Более тяжело болеют мальчики, дебют, как правило, в раннем возрасте. Мышечная слабость начинается с мимических мышц (неспособность плотно зажмурить глаза или сжать губы). За этим следует клиническая картина слабости лопаточных мышц с отстоянием (крыловидностью) лопатки и затруднением отведения плеча. Также может быть слабость передних большеберцовых мышц.
Состояние вызвано делецией длинного плеча 4 хромосомы; подтверждает диагноз генетическое тестирование, являющееся очень чувствительным и специфичным.
- Читать далее "Симптомы миотонии и ее лечение"
Оглавление темы "Поражения нервной системы":- Симптомы периферических нейропатий - полинейропатии
- Симптомы наследственной нейропатии
- Симптомы нейропатии из-за нарушения обмена веществ (метаболическая нейропатия)
- Симптомы нейропатии при инфекции
- Оценка боли и ее восприятия
- Симптомы фибромиалгии и ее лечение
- Симптомы артрогриппоза и его лечение
- Симптомы мышечной дистрофии и ее лечение
- Симптомы миотонии и ее лечение
- Классификация периферических нейропатий и их причины