
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
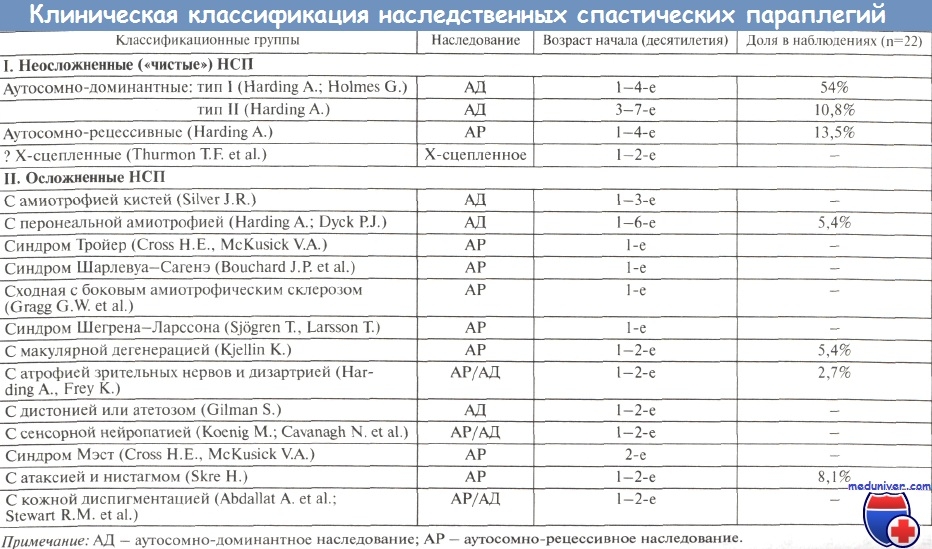
Варианты Х-сцепленных осложненных спастических параплегий
В данной группе практически все заболевания наследуются по Х-сцепленному рецессивному типу, и лишь при одной форме имеет место Х-сцепленное доминантное наследование болезни.
Синдром Пэйна (микроцефалия со спастической диплегией). Синдром Пэйна (MIM 311400) описан в 1960 г. во франкоязычной канадской семье (Paine R.S.). Помимо микроцефалии и спастического парапареза у больных наблюдались миоклонические припадки и изменение содержания аминокислот в цереброспинальной жидкости, а в секционном случае обнаружены пороки развития головного мозга (гипоплазия мозжечка, нижних олив и моста). В последующих наблюдениях содержание аминокислот в цереброспинальной жидкости было нормальным.
E. Seemanova и соавт. в семье с 6 больными выделили вариант синдрома Пэйна с отсутствием брюшных рефлексов, нормальной цереброспинальной жидкостью и без пороков развития мозга (синдром Семановой). Некоторые авторы разделяют синдромы Пэйна и Семановой, но в каталоге MIM они объединены. В некоторых более ранних руководствах синдром Пэйна описывался в группе врожденных гипоплазии мозжечка (Harding А.Е.).
Спастическая параплегия с умственной отсталостью и ладонно-подошвенным гиперкератозом, синдром Фицсиммонса (MIM 309560). Это сочетание имелось у 4 братьев 16—35 лет в семье, описанной J.Fitzsimmons и соавт.. У троих спастический парапарез с деформацией стоп был выраженным, у четвертого больного деформация была негрубой, а парапарез проявлялся только гиперрефлексией; у матери имелся только подошвенный гиперкератоз, а три сестры 28—34 лет были полностью здоровы.
Спастическая параплегия с синдромом Кальмана (MIM 308750). Синдром Кальмана — Х-сцепленное заболевание, проявляющееся гипогонадотропным гипогонадизмом и аносмией (MIM 308700). Его сочетание со спастической параплегией описано у двух братьев (Tuck R. et al.).
Имелись дополнительные неврологические симптомы (расстройства чувствительности и односторонняя наружная офтальмоплегия), а у одного из братьев — нарушение цветового зрения. У сестры обнаружена легкая неврологическая симптоматика, у матери - негрубые изменения ЭМГ, связь которых с болезнью братьев не доказана. Авторы предположили Х-сцепленное либо ограниченное полом доминатное наследование спастической параплегии в данной семье, независимое от синдрома Кальмана. Наличие особого синдрома сомнительно, так как брат матери страдал гипогонадизмом без неврологических симптомов.
Спастическая параплегия с глухотой (MIM 312910) имелась в семье с 6 больными мужчинами в трех поколениях (Wells С, Jancovic J.). Спастический парапарез и нейросенсорная глухота начинались примерно в Шлет; у больных наблюдались также непостоянные сопутствующие симптомы: тремор, катаракта, пигментная дегенерация сетчатки, гипогонадизм, низкорослость.
Спастическая параплегия с поражением белого вещества мозга описана у 5 мужчин в одном поколении семьи (Gutmann D. et al.). Помимо спастического парапареза болезнь включала умственную отсталость, дизартрию, атаксию и тремор. Неврологические нарушения появлялись на 1-м десятилетии жизни, нарастали в течение 3—4 лет, затем течение было стационарным. При МРТ обнаруживались изменения белого вещества в перивентрикулярных областях. У 3 из 4 обследованных больных имелись также расстройства цветового зрения.
Младенческая НСП с нистагмом наблюдалась в семье с 5 больными (Steinmuller R. et al.). Неврологические расстройства начинались до 3 мес. и включали в себя нистагм (первый симптом, впоследствии исчезавший), задержку двигательного развития, нарастающий нижний спастический парапарез с отсутствием ходьбы, затем спастичность в руках, снижение зрения, расстройства речи (моторная афазия); умственная отсталость при этом была легкой.
Тяжелая раннелетальная НСП описана K.-H.Gustavson и соавт. (1993). Помимо спастической параплегии болезнь включала глубокую умственную отсталость, тяжелое поражение глаз, глухоту, эпилепсию и контрактуры суставов.
НСП с гипоплазией нижней челюсти имелась у 4 больных в бельгийской семье, описанной S.Claes и соавт.. В этой семье спастический парапарез прогрессировал медленно, а у больных наряду с нарушением развития нижней челюсти отмечалась также тяжелая умственная отсталость.
Х-сцепленная доминантая НСП, летальная у мужчин (единственный пример такого наследования среди НСП), предположена G. Woods и соавт. в семье, где женщины страдали начинавшимся в детстве прогрессирующим спастическим парапарезом с нарушением тазовых функций, а новорожденные мальчики погибали на фоне гипотонии и дыхательной недостаточности. У женщин, кроме того отмечены непрогрессирующее снижение сумеречного зрения (при нормальной электроретинограмме) и снижение уровня иммуноглобулинов G2 без клинических проявлений. Ген предположительно картирован в локусе Xq26-qter (Woods G. et al.).
В наших популяционных исследованиях и практике медико-генетического консультирования не встретились НСП с несомненным Х-сцепленным наследованием, хотя его нельзя полностью исключить в несемейных случаях у мужчин и в тех семьях, где больны только братья.
Вероятно, далеко не все клинические варианты генетически самостоятельны. Определить их место в широком спектре НСП помогут дальнейшие исследования, прежде всего молекулярно-генетические.
- Читать "Лекарства для лечения наследственных спастических параплегий (НСП)"
Оглавление темы "Осложненные наследственные спастические параплегии (НСП)":- Спастическая параплегия с аномальным строением метафизов и поражением кожи
- Редкие формы рецессивных наследственных спастических параплегий (НСП)
- Эпидемиология рецессивных наследственных спастических параплегий (НСП)
- Варианты Х-сцепленных осложненных спастических параплегий
- Лекарства для лечения наследственных спастических параплегий (НСП)
- Физиотерапия и операции в лечении наследственных спастических параплегий (НСП)
- Медико-генетическое консультирование при наследственных спастических параплегий (НСП). Профилактика
- Вывод из запоя: срочно и круглосуточно