
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Трансгенные модели атаксии Фридрейха
В основе атаксии Фридрейха лежит количественная и функциональная недостаточность митохондриального белка фратаксина в клетках и тканях. Фратаксин является эволюционно консервативным белком, и его гомолог обнаруживается у грамотрицательных бактерий, дрожжей, а также мышей, собак и других млекопитающих (Pandolfo М., Knight S. et al., Kuiper Н. et al.).
Наиболее сохранным у эволюционно дистантных организмов является участок белка протяженностью 27 аминокислот, кодируемый экзонами 4 и 5а (Puccio Н. et al.). Исследования с использованием технологии гибридизации in situ показали, что экспрессия фратаксина у мышей близка к таковой у человека и хорошо коррелируете основными «мишенями» нейродегенерации при атаксии Фридрейха (Koutnikova Н. et al.).
После получения этих данных в ведущих лабораториях мира началось активное изучение-функции гомологов фратаксина на различных экспериментальных моделях (Pandolfo М., Adinolfi S. et al.).
Значительная роль исследований дрожжевых и бактериальных клеток в раскрытии патофизиологических основ атаксии Фридрейха была представлена в предыдущих разделах главы. В частности, Н. Koutnikova и соавт. показали, что инактивация гомолога фратаксинового гена у дрожжей сопровождается глубокими нарушениями функции митохондрий в данной клеточной системе. В настоящее время основное внимание уделяется моделированию атаксии Фридрейха на высших позвоночных животных.
C.J.Miranda и соавт. создали линию трансгенных мышей, у которых в их собственный ген Frda (гомолог человеческого гена FRDA) встроен экспандированный участок с увеличенным числом копий GAA-повторов. У этих животных уровень фратаксина в тканях составил 25—36% от нормы, что, однако, не сопровождалось развитием каких-либо двигательных нарушений, накоплением железа в клетке или мейотической/митотической нестабильностью мутантного гена.
В большой серии работ был использован иной подход к созданию трансгенных моделей атаксии Фридрейха — «нокаут» (инактивация) у животных гомолога человеческого гена Frda. Так, в 2002 г. M.Cossee и соавт. создали трансгенную линию мышей с полностью инактивированным фратаксином путем моделирования в геноме животных делеции 4-го экзона гена Frda.
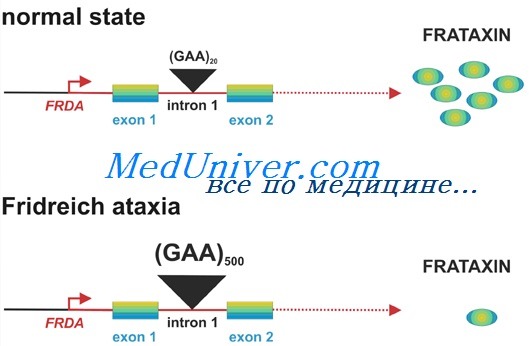
Гомозиготная делеция данного гена приводила к ранней эмбриональной летальности уже спустя несколько дней после имплантации, что доказывает эссенциальную, жизненно важную роль фратаксина в функционировании клетки; при этом в тканях элиминированного эмбриона не было выявлено аккумуляции железа, по-видимому, в связи с тем, что клеточная гибель является следствием самостоятельного, Fe-независимого механизма (Cossee М. et al.).
Для создания адекватной трансгенной модели атаксии Фридрейха и преодоления вышеуказанного летального эффекта «полного нокаута» гена Frda в работе Н.Puccio и соавт. был использован так называемый «кондиционный нокаут». В этом случае с помощью специальной технологии мечения встраиваемых трансгенных конструкций достигается адресная избирательная инактивация гена в определенной ткани в определенный момент времени.
Используя эту технологию, авторам удалось создать 2 линии трансгенных мышей: одну с дефицитом фратаксина в поперечнополосатой мускулатуре, а другую - в нейронах мозга и сердечной мышце (Puccio Н. et al., 2001). Эти модели воспроизвели ряд важнейших прогрессирующих патофизиологических и биохимических характеристик атаксии Фридрейха, таких как гипертрофия миокарда, дисфункция крупных чувствительных нейронов спинномозговых ганглиев, дефицит активности аконитазы и комплексов I—III дыхательной цепи митохондрий, а также внутримитохондриальное накопление железа, начинающееся после инактивации Fe-S-зависимых ферментов.
В 2004 г. была опубликована работа, в которой авторы на основе использования аналогичного «кондиционного» подхода к экспрессии трансгена представили результаты создания линий мышей с прогрессирующей смешанной сенсорно-мозжечковой атаксией - главным симптомом атаксии Фридрейха (Simon D. et al.).
Гистологически у животных отмечалась патология спинного мозга и спинномозговых чувствительных ганглиев с отсутствием моторной невропатии, что весьма близко напоминает морфологическое «ядро» атаксии Фридрейха; у животных имелись также дефекты арборизации клеток Пуркинье и гибель гранулярных клеток коры мозжечка. Авторы выявили процесс аутофагии в дорсальных спинно-мозговых ганглиях животных, сопровождающийся удалением митохондриального детрита и появлением отложений липофусцина.
Полученные линии мышей с «кондиционным нокаутом» фратаксинового гена являются первыми моделями атаксии Фридрейха с медленным прогрессированием процесса нейродегенераиии, созданными у млекопитающих. Это крайне важно для детального изучения патологического каскада и тестирования новых стратегий лечения болезни. Предварительные результаты исследования эффективности новых лекарственных препаратов (в частности идебенона) у таких трансгенных животных (Seznec H.et al.) подробнее рассмотрены далее в статьях на нашем сайте.
- Читать "Критерии диагностики атаксии Фридрейха"
Оглавление темы "Клиника атаксии Фридрейха":- Биохимические анализы при атаксии Фридрейха
- Морфология и гистохимия мышц при атаксии Фридрейха
- Трансгенные модели атаксии Фридрейха
- Критерии диагностики атаксии Фридрейха
- Неврологические симптомы атаксии Фридрейха
- Болезни сердца при атаксии Фридрейха
- Эндокринные нарушения при атаксии Фридрейха. Причины диабета
- Костные деформации при атаксии Фридрейха
- Пример атаксии Фридрейха классического фенотипа
- Атипичные варианты атаксии Фридрейха