
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Спастическая параплегия 3-го типа (SPG3 или SPG3A) - клиника, диагностика
С 1986 г., когда был картирован ген SPG1, молекулярная генетика сделала большой шаг вперед: в настоящее время картировано почти 30 локусов наследственной спастической параплегии (НСП). Достаточно высокими темпами идет и клонирование соответствующих мутантных генов. Особенно резкий рывок произошел в самое последнее время — в частности сгладился имевшийся разрыв между значительным количеством известных хромосомных локусов и сравнительно небольшим числом идентифицированных генов.
Большинство аутосомно-доминантных НСП с известными локусами относят к неосложненным формам.
Спастическая параплегия-3 (MIM 182600) встречается повсеместно. По одним данным, она составляет всего 6—10% доминантных наследственной спастической параплегий (Fink J.et al., Zhao X. et al., Wilkinson P. et al.), подругам — занимает значительное место в структуре доминантных форм, возможно, второе после SPG4 (Durr A. et al., Sauter S. et al.).
Клиническая картина — типичная для неосложненных форм. Начало болезни преимущественно раннее: из 4 семей, анализируемых J. Fink, в 3 возраст начала составил 2-15 лет (в основном до 7 лет) и лишь в одной семье варьировал от 2 до 50 лет. У всех больных из 5 семей, описанных X. Zhao и соавт., болезнь началась до 10 лет, а у большинства из них — до 5 лет; в 12 французских семьях средний возраст начала составил 4,6+3,9 года (Diirr A. et al.), в итальянской семье — 8 лет (Muglia М. et al.); F. Dalpozzo и соавт. наблюдали начало SPG3 и младенчестве.
Вместе с тем возможен и поздний дебют болезни (Sauter S. et al.). Прогрессирование, как правило, медленное. Вначале сообщалось о большей тяжести SPG3 по сравнению с другими доминантными НСП, но сейчас это не подтверждается: в больших выборках пациентов доля тяжелых случаев SPG3 такая же, как при других формах (Gispert S. et al., Huang S. et al.). Реже, чем при других доминантных НСП, при SPG3 наблюдаются гиперрефлексия в руках, расстройства тазовых функций и вибрационной чувствительности в поздней стадии болезни, тогда как сколиоз и амиотрофия голеней — напротив, встречаются чаще (Diirr A. et al.). В части семей с SPG3 отмечена антиципация (Thurmon F. et al.). В семьях значительна доля субклинических случаев болезни.
SPG3 — первый картированный локус из всех доминантных НСП (Hazan J. et al., Gispert S. et al.). Ген заболевания локализован в хромосомной области 14q11.2—21. Второе обозначение данного локуса, SPG3A, дано для отграничения от клинически идентичной НСП, при которой не обнаруживается сцепление с хромосомой 14q.
Практическим выходом картирования локуса SPG3 стала косвенная пренатальная ДНК-диагностика в одной из семей (Hedera Р. et al.).
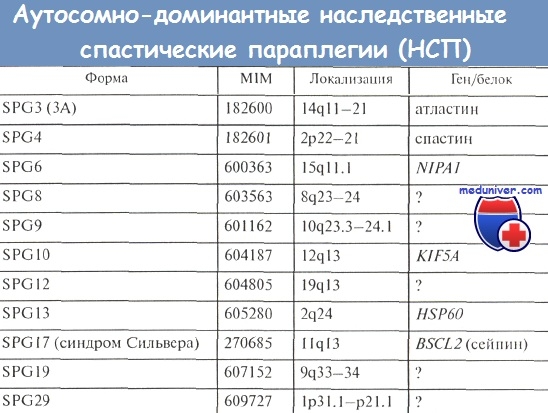
В 2001 г. был идентифицирован ген болезни (Zhao X. et al.). Он кодирует новый белок атластин, состоящий из 588 аминокислот. Атластин экспрессируется достаточно широко, но преимущественно в головном и спинном мозге. Не имея гомологии с другими известными молекулярными продуктами наследственной спастической параплегии, атластин вместе с тем обнаружил значительную гомологию с несколькими крупными ГТФазами, особенно с гуанилатсвязывающим белком 1 (GBP-1).
Эти ГТФазы принадлежат к семейству динаминов — белков, участвующих в синаптическом транспорте и дисперсии митохондрий, а также способных связываться с микротрубочками нейронального цитоскелета (Zhao X. et al., Zhu P. et al.).
Первооткрыватели гена X. Zhao и соавт. обнаружили специфичные, связанные с болезнью миссенс-мутации атластина в 5 семьях с SPG3, еще 2 миссенс-мутации найдены в итальянских семьях (Muglia М. et al., Tessa A. et al.); одна из мутаций, R239C, встретилась в 3 неродственных семьях. Как уже указывалось, относительно распространенности SPG3 есть разные данные.
Например, P. Wilkinson и соавт. обнаружили мутацию атластина (ту же R239C) только в одной из 12 семей с ранней неосложненной доминантной НСП и предварительно исключенной формой SPG4, что говорит о редкости SPG3. В то же время, S.Sauter и соавт. обследовали 13 семей с доминантной наследственной спастической параплегии (тоже вначале исключив SPG4, но не ограничиваясь ранним началом болезни) и нашли мутации атластина в 5 семьях (38%); 2 мутации были описаны впервые, причем одна из них встретилась в двух неродственных семьях с разным возрастом начала болезни — типичным ранним и атипичным поздним.
Сходные данные получены A. Durr и соавт. (2004), которые обследовали на предмет мутаций атластина 31 французскую семью с неосложненной доминантной наследственной спастической параплегии (после исключения SPG4). В 12 (39%) семьях выявлено 9 разных миссенс-мутаций, в том числе 7 впервые описанных. Из 34 носителей мутации 7 (21%) считали себя здоровыми, причем у двух из них отсутствовали и объективные симптомы, что указывает на неполную пенетрантность гена.
- Читать "Спастическая параплегия 4-го типа (SPG4) - клиника, диагностика"
Оглавление темы "Наследственные спастические параплегии (НСП)":- Морфология наследственной спастической параплегии (НСП)
- Диагностика наследственной спастической параплегии (НСП)
- Дифференциальная диагностика наследственной спастической параплегии (НСП)
- Спастическая параплегия 3-го типа (SPG3 или SPG3A) - клиника, диагностика
- Спастическая параплегия 4-го типа (SPG4) - клиника, диагностика
- Пример спастической параплегии 4-го типа (SPG4)
- Пример ДНК-диагностики спастической параплегии 4-го типа (SPG4)
- Спастическая параплегия 6-го типа (SPG6) - клиника, диагностика
- Спастическая параплегия 8-го типа (SPG8) - клиника, диагностика
- Спастическая параплегия 9-го типа (SPG9) - клиника, диагностика