
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Спастическая параплегия 2-го типа (SPG2) - клиника, диагностика
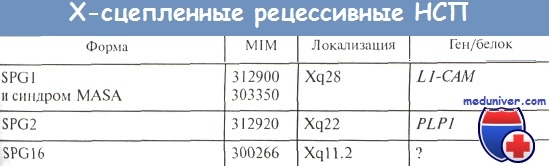
Форма SPG2 (MIM 312900), как правило, манифестирует до 20 лет, чаще в раннем детстве; случаи позднего начала болезни единичны (Sivakumar К. et al.). Более типичны неосложненные формы, и в каталоге MIM форма SPG2 отнесена к неосложненным НСП, но некоторые авторы включают ее в число осложненных вариантов параплегии (Fink J.). Действительно, встречаются оба варианта фенотипов.
В первой семье, где был картирован ген болезни (Keppen L. et al.), у 12 больных имелась неосложненная форма НСП с началом в раннем детстве. Описаны и другие семьи с неосложненной НСП, нормальной картиной МРТ и обычной продолжительностью жизни. Вместе с тем, в ряде клинически неосложненных случаев при МРТ обнаружены изменения белого вещества (Cambi F. et al.), а некоторые семьи демонстрируют целый комплекс дополнительных симптомов и гораздо более тяжелое течение.
Так, в семье, описанной A. Johnston и V. McKusick еще в 1962 г. и позже обследованной с помощью молекулярно-генетических методов (Kobayashi Н. et al.), болезнь манифестировала с возраста становления ходьбы как неосложненная НСП, но позже присоединялись нистагм, дизартрия, расстройства чувствительности, деменция и атрофия зрительных нервов (в разных сочетаниях), а затем — атрофия мышц и контрактуры; уже в молодости утрачивалась возможность самостоятельной ходьбы.
Осложненный и неосложненный варианты могут сочетаться внутрисемейно: в семье, описанной D.Bonneau и соавт., у одних больных была неосложненная НСП, у других параплегия в сочетании с олигофренией.
Возможны легкие или умеренные проявления гетерозиготного носительства у женщин — клинические и МР-томографические (Cambi F. et al., Sivakumar К. et al., Inoue K. et al.).
В критическом хромосомном участке Xq22 еще в 1985 г. был картирован ген протеолипидного белка PLP1 (Willard Н., Riordan J.), а 2 года спустя установлена связь с этим локусом Х-сцспленной НСП типа SPG2 (Keppen L. et al.). Продукт гена PLP1 -липофилин, главный компонент миелина ЦНС (Saugier-Veber P. et al.). Липофилин имеет две изоформы (точнее, два продукта альтернативного сплайсинга гена PLP1): протеолипидный белок PLP, необходимый для компактной укладки миелина, и белок DM20, необходимый для развития олигодендроцитов (Griffiths I. et al.).
Различные мутации PLPI вызывают не только форму спастической параплегии SPG2, но и гораздо более тяжелое многосимптомное заболевание из группы лейкодистрофий детского возраста - болезнь Пелицеуса-Мерцбахера (М1М 312080). Болезнь Пелицеуса-Мерцбахера характеризуется врожденной гипотонией, нистагмом, задержкой психического развития, прогрессирующими пирамидными, мозжечковыми и дистоническими нарушениями. При SPG2 повреждается только белок PLP, а белок DM20 не изменен - в отличие от болезни Пелицеуса-Мерцбахера, при которой страдают оба белка.
Показано, что дупликации и другие мутации, влияющие на роль белковых продуктов гена в отношении созревания олигодендроцитов, проявляются тяжелым фенотипом болезни Пелицеуса-Мерцбахера; в то же время, мутации, затрагивающие лишь процессы компактизации миелина, приводят к манифестации более легких аллельных заболеваний - «чистой» и осложненной форм спастической параплегии (Reid Е.).
Таким образом, фенотип в каждом конкретном случае зависит от мутации: несколько миссенс-мутаций вызывают SPG2, тогда как болезнь Пелицеуса-Мерцбахера связана чаще всего с дупликациями всего гена. При болезни Пелицеуса-Мерцбахсра описаны также полные дслсции и трипликации гена (Saugier-Veber P. et al., Sivakumar К. et al., Yool D. et al.); самые же тяжелые формы болезни Пелицеуса-Мерцбахсра возникают при точковых мутациях в наиболее консервативных аминокислотных последовательностях DM20 (Cailloux F. et al.).
Межсемейное клиническое разнообразие SPG2 также связано с характером мутации, но, очевидно, и с другими причинами, поскольку существуют значительные внутрисемейные различия фенотипов болезни.
Точный механизм развития параплегии вследствие мутаций PLP1 не установлен. В некоторых семьях с SPG2 при МРТ обнаружены изменения белого вещества ЦНС, характерные для демиелинизирующих заболеваний, в том числе для болезни Пелицеуса—Мерцбахера (Cambi F. et al.). Сходные изменения в веществе мозга имеются у мышей-«трясогузок» {rumpshaker), представляющих собой природную модель SPG2: у них снижено содержание белка PLP в ЦНС, а содержание DM20 не изменено (Kobayashi Н. et al.).
Предполагаемые механизмы спастичности при SPG2 — нарушение миелинизации пирамидных путей, а также повреждение оли-годендроцитов в результате нарушенного транспорта белков (Yool D. et al.).
При SPG2 имеется практический опыт прямой ДНК-диагностики, включая пренатальную диагностику носительства мутации у плода.
Особняком стоит наблюдение J.Arena и соавт.. Х-сцепленный синдром в наблюдавшейся ими семье включает тяжелый нижний спастический парапарез (больные никогда не ходили) с минимальным вовлечением рук, умственную отсталость и необычные изменения МРТ: наряду с макрогирией и гипомиелинизацией отмечено резкое повышение парамагнитного сигнала с подкорковых ядер, свидетельствующее об отложении железа (в отсутствие экстрапирамидных симптомов). Выявлено сцепление с болезни с локусом Xq22—25. Хотя не исключено перекрывание с локусом SPG2, налицо заметные клинические отличия данного синдрома даже от осложненных случаев SPG2. Нозологическая принадлежность этого наблюдения не уточнена.
- Читать "Спастическая параплегия 16-го типа (SPG16) - клиника, диагностика"
Оглавление темы "Наследственные спастические параплегии (НСП)":- Спастическая параплегия 1-го типа (SPG1) - клиника, диагностика
- Спастическая параплегия 2-го типа (SPG2) - клиника, диагностика
- Спастическая параплегия 16-го типа (SPG16) - клиника, диагностика
- Синдром Шегрена-Ларссона - клиника, диагностика
- Пример синдрома Шегрена-Ларссона
- Спастическая атаксия типа Шарлевуа-Сагенэ - клиника, диагностика
- Ранний восходящий спастический паралич и ювенильный первичный боковой склероз - клиника, диагностика
- Нарушение распознавания клеток и клеточных сигналов L1-CAM при НСП
- Нарушение процессов миелинизации при НСП: протеолипидный белок и его изоформы
- Нарушения в системе митохондриальных молекулярных шаперонов при НСП и HSP60