
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Синдром Шегрена-Ларссона - клиника, диагностика
Синдром Шегрена-Ларссона (MIM 279200) - редкое аутосомно-рецессивное заболевание, описанное в Швеции (Sjogren Т., Sjogren Т., Larsson Т.), где имеется его накопление: средняя распространенность болезни в стране за 1901-1977 гг. составляет 0,6 на 100 000 населения, а в одном из северо-восточных графств достигает 10,2 на 100 000.
Все эти случаи связывают с мутацией, возникшей около 600 лет назад (Jagell S. et al.). Однако синдром Шегрена-Ларссона не является исключительно «шведской» болезнью и, будучи редким, встречается повсеместно: в странах Запада — Германии, Бельгии, Голландии, Испании, - в США (в том числе у индейцев), Канаде, в странах Ближнего Востока, Японии (Rogers G. et al., Pigg M. et al., Aoki N. et al.), а также в России, что подтверждает наше наблюдение (Рудснская Г.Е., Журкова Н.В.).
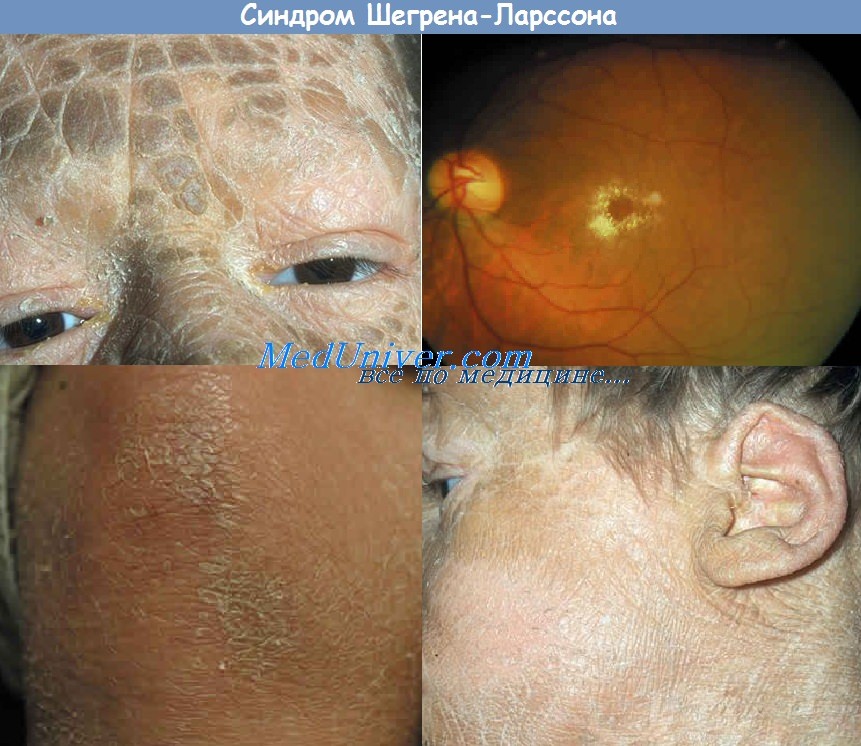
Основные симптомы - врожденный ихтиоз, олигофрения и спастический тетрапарез (преимущественно нижний парапарез). Ихтиоз при синдроме Шегрена-Ларссона неотличим от врожденной ихтиозиформной эритродермии, которая чаще встречается как самостоятельное рецессивное заболевание. Как правило, ихтиоз имеется уже с рождения, носит распространенный характер, особенно выражен на нижней части живота, шее, спине, сгибательных поверхностях рук и ног, меньше — на лице, ладонях и подошвах, иногда сопровождается зудом.
Эритема отчетлива в младенчестве, с возрастом исчезает; потоотделение и придатки кожи не страдают. Тяжесть ихтиоза варьирует, но не нарастает, а нередко с возрастом уменьшается. Патоморфология кожи та же, что при всякой ихтиозиформной патологии, специфичных признаков нет. Психоневрологические расстройства становятся явными к году. Большинство больных самостоятельно не ходят из-за спастического пареза ног, парез рук выражен гораздо меньше. Двигательные расстройства могут умеренно нарастать (до пубертатного периода) либо имеют стационарное течение, напоминая клинику ДЦП. Умственная отсталость (в степени выраженной дебильности или имбецильности) не нарастает, у 30-50% больных наблюдаются эпилептические припадки.

Нейровизуализация обнаруживает изменения белого вещества разной степени и локализации — очаги в псривентрикулярных областях либо множественные, диффузно разбросанные в белом веществе очаги с тенденцией к слиянию. Непостоянный, но патогномонич-ный ранний признак (особенно частый у шведских больных) — белесые блестящие пятнышки на сетчатке; у половины больных имеется пигментная дегенерация сетчатки.
Еще один факультативный симптом — умеренная низкорослость. Продолжительность жизни достаточно велика, описаны больные старше 50 лет.
Синдром Шегрена—Ларссона традиционно включают в число осложненных форм НСП (Иванова-Смоленская И.А. с соавт., Harding А.), но в конце 1980-х годов установлена его принадлежность к наследственным нарушениям обмена липопротеинов.
Биохимический дефект связан со снижением активности мембраносвязанной дегидрогеназы жирных альдегидов (FALDH: Fatty Aldehyde Dehydrogenase) — компонента комплекса «жирный спирт: НАД+оксиредуктаза» (Rizzo W. et al.). В кожных фибробластах и лейкоцитах периферической крови резко (до 10% от нормальных показателей) снижены активность FALDH и всего комплекса «жирный спирт: НАД+оксиредуктаза»; вторичный биохимический маркер болезни — снижение концентрации гексадсканола.
Эти нарушения обнаруживаются уже у плода, т.е. возможна биохимическая пренатальная диагностика. Биохимические изменения (в среднем на 50% от нормы) выявляют и у гетерозиготных носителей мутантного гена. Имеются единичные наблюдения «неполного» синдрома Шегрена—Ларссона с редуцированной клинической картиной и частичным дефицитом фермента (Kawakami Т. et al.).
Картирование (хромосома 17р11.2) и идентификация гена FALDH проведены в шведских семьях, а последующие исследования семей разной этнической принадлежности показали генетическую однородность заболевания (Rogers G. et al., De Laurenzi V. et al., Pigg M. et al., Rizzo W. et al., Aoki N. et al.).
В настоящее время при синдроме Шегрена—Ларссона описаны десятки разнообразных мутаций FALDH, некоторые из них встретились в единичных семьях, другие — многократно (в частности мутация, преобладающая в Швеции). Стала возможной практическая ДНК-диагностика болезни, в том числе пренатальная.
Лечения, помимо симптоматического, не существует. Попытки диетотерапии (исключение или ограничение продуктов, содержащих жирные кислоты с длинной цепью, и замена их жирными кислотами с цепью средней длины) не дали убедительного эффекта.
- Читать "Пример синдрома Шегрена-Ларссона"
Оглавление темы "Наследственные спастические параплегии (НСП)":- Спастическая параплегия 1-го типа (SPG1) - клиника, диагностика
- Спастическая параплегия 2-го типа (SPG2) - клиника, диагностика
- Спастическая параплегия 16-го типа (SPG16) - клиника, диагностика
- Синдром Шегрена-Ларссона - клиника, диагностика
- Пример синдрома Шегрена-Ларссона
- Спастическая атаксия типа Шарлевуа-Сагенэ - клиника, диагностика
- Ранний восходящий спастический паралич и ювенильный первичный боковой склероз - клиника, диагностика
- Нарушение распознавания клеток и клеточных сигналов L1-CAM при НСП
- Нарушение процессов миелинизации при НСП: протеолипидный белок и его изоформы
- Нарушения в системе митохондриальных молекулярных шаперонов при НСП и HSP60