
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Синдром MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes) - клиника, диагностика
Синдром MELAS (MIM 540000) обозначается по первым буквам английского названия болезни «Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes» (митохондриальная энцефаломиопатия, лактат-ацидоз и инсультоподобные эпизоды).
Синдром MELAS обычно манифестирует в возрасте от 5 до 20 лет. Он проявляется в первую очередь острыми инсультоподобными эпизодами с развитием очаговых изменений в затылочной и теменно-височной областях мозга и появлением соответствующей неврологической симптоматики (парезы, корковые расстройства зрения, судороги, кома, приступы головной боли и рвоты и др.).
Появление очагов связывают с преходящей дисфункцией окислительного фосфорилирования в паренхиме мозга, а также структурно-метаболическими нарушениями в стенках артериол и капилляров; характерной особенностью таких «смешанных» по генезу инфарктов мозга является относительно быстрое восстановление, что можно проследить при КТ/МРТ-исследовании.
Атаксия может быть заметным симптомом болезни у некоторых пациентов с синдромом MELAS (Bertini Е. et al.). При синдроме MELAS могут наблюдаться также миопатические проявления (повышенная утомляемость и непереносимость физических нагрузок), демснция, дегенерация сетчатки, нейросенсорная глухота, низкорослость, диабет, кардиомиопатия и целый ряд других мультиорганных проявлений. Характерен значительный уровень лактат-ацидоза в крови и цереброспинальной жидкости, при биопсии скелетных мышц нередко выявляется феномен «рваных красных волокон».
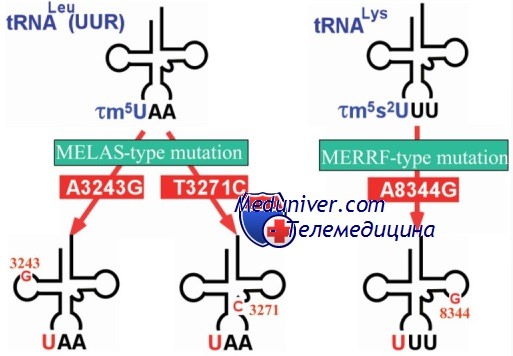
Синдром MELAS наследуется по материнскому типу, однако исключительная вариабельность клинических проявлений может весьма затруднять оценку семейного анамнеза. У больных с синдромом MELAS описано как минимум 8 точковых мутаций в генах мтДНК., причем 5 из них локализованы в различных участках гена тРНКLeu (UUR)) (DiMauro S., Chinnery P. et al.). Наиболее частой мутацией является замена A-»G в положении 3243 (около 80% больных), а в целом мутации указанного гена лейциновой тРНК обнаруживаются почти в 95% случаев MELAS.
В редких случаях у больных с синдромом MELAS описаны точковые мутации в генах других тРНК и гене СОХ III-субъединицы IV комплекса дыхательной цепи. Все мутации обнаруживаются в гетероплазмическом состоянии.
- Читать "Синдром NARP (Neuropathy, Ataxia, Retinitis Pigmentosa) - клиника, диагностика"
Оглавление темы "Атаксии - клиника, диагностика":- Эпизодическая атаксия с пароксизмальным хореоатетозом и спастичностью - клиника, диагностика, лечение
- Аутосомно-доминантная спастическая атаксия (синдром SAX1) - клиника, диагностика
- Аутосомно-доминантная спастическая атаксия с умственной отсталостью (синдром SPAR) - клиника, диагностика
- Заднестолбовая атаксия - клиника, диагностика
- Синдром Герстманна-Штреуслера—Шейнкера - клиника, диагностика
- Наследственные метаболические атаксии - определение, классификация
- Митохондриальные атаксии - клиника, диагностика
- Синдром MERRF (Myoclonus Epilepsy with Ragged-Red Fibers) - клиника, диагностика
- Синдром MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes) - клиника, диагностика
- Синдром NARP (Neuropathy, Ataxia, Retinitis Pigmentosa) - клиника, диагностика