
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Клиника и патогенез синдрома FXTAS
Синдром FXTAS (синдром атаксии/тремора, ассоциированный с фрагильной Х-хромосомой) — возрастзависимое нейродегенеративное заболевание, описанное R.J.Hagerman и соавт. в 2001 г. Оно может развиваться у пожилых лиц мужского пола, являющихся носителями «премутантного» аллеля гена FMR1 (т.е. аллеля с числом тандемных CGG-повторов от 50—60 до 200 копий, не связанного непосредственно с детской умственной отсталостью типа FRAXA).
Предварительные оценки частоты этого нового синдрома были получены при анализе больших семей с умственной отсталостью типа FRAXA, в которых оказалось возможным тотально обследовать родственников больных детей по женской линии (т.е. носителей мутантной хромосомы с различной степенью экспансии CGG-повторов). Оказалось, что вполне определенная форма нейродегенерации развивается не менее чем у 1/3 мужчин старше 50 лет, являющихся носителями «премутации» в гене FMRI (Jacquermont S. et al.).
Есть основания полагать, что в более пожилой возрастной группе пенетрантность «премутантного» аллеля FMR1 может быть еще выше — вплоть до 75% у носителей-мужчин старше 80 лет (Hagerman P.J., Hagerman R.J., Jacquemont S. et al.).
Клиническая картина синдрома FXTAS достаточно вариабельна. Чаше всего при этом заболевании у мужчин в возрасте 50—70 лет развиваются прогрессирующий интенционный тремор, нарастающие расстройства ходьбы атактического типа, дизметрия и мозжечковая дизартрия. Очень характерно, что значительная выраженность интенционного тремора не соответствует более «мягким» проявлениям других мозжечковых симптомов; в 10-30% случаев имеется тремор покоя, а также тремор эссенциального либо смешанного типа.
Примерно у 60% больных синдромом FXTAS развивается паркинсонизм (брадикинезия, ригидность, реже паркинсоновское дрожание), который обычно выражен в умеренной степени и не чувствителен к препаратам леводопы. Весьма типичны также вегетативные расстройства (недержание мочи и кала, импотенция). В ряде случаев в структуру синдрома FXTAS могут входить нарушения памяти и дефицит экзекутивных функций, постепенно приводящие к когнитивному снижению; может наблюдаться периферическая невропатия со снижением вибрационной и тактильной чувствительности в дистальных отделах конечностей, выпадением ахилловых рефлексов; у части больных отмечаются слабость в проксимальных отделах ног, жгучие боли либо онемение в ногах и судороги типа крампи; в некоторых случаях развиваются психиатрические симптомы — тревожность, депрессия и т.п.
Хотя у абсолютного большинства женщин-носительниц синдром FXTAS не развивается (как и следует ожидать при Х-сцепленном рецессивном наследовании), в литературе все же описаны несколько случаев этого синдрома у женщин, имеющих типичный «промежуточный» аллель FMR1 с числом CGG-повторов от 78 до 93 (Hagerman R.J. et al.). Характерной клинической особенностью у большинства наблюдавшихся женщин было сравнительно «мягкое» течение синдрома и меньшая тяжесть клинических проявлений по сравнению с мужчинами (так, ни у кого из женщин с синдромом FXTAS не было отмечено значимых когнитивных нарушений). Предполагается, что редкие примеры синдрома FXTAS у женщин могут быть связаны с несбалансированной лайонизацией (преимущественной инактивацией нормальной Х-хромосомы), нарушением имеющегося в норме протективного эффекта эстрогенов и другими факторами (Hagerman RJ. et al.).

В единичных исследованных секционных случаях синдрома FXTAS (втом числе у женщин) выявлены генерализованная атрофия мозжечка и больших полушарий мозга, спонгиоз белого вещества и средних мозжечковых ножек, гибель клеток Пуркинье. Наиболее значимой находкой стали эозинофильные внутриядерные включения в нейронах и астроцитах, широко распространенные в различных отделах ЦНС — полушарной коре (особенно в лобных отделах), стволе мозга, гиппокампе (Greco C.М. et al., Hagerman R.J. et al.).
Эти включения убиквитин-позитивны, что является универсальным признаком большого числа нейродегенертаивных заболеваний человека и свидетельствует о нарушении при FXTAS клеточных механизмов убиквитин-протеасомной деградации аномальных белков (Иллариошкин С.Н., Hagerman P.J., Hagerman R.J.). В то же время, отмеченные включения существенно отличаются от всех других известных включений отсутствием положительной реакции на а-синуклеин, белок тау либо полиглутаминовый эпитоп, что принципиально отличает FXTAS от болезни Паркинсона, множественной системной атрофии, таупатий и полиглутаминовых болезней. Таким образом, синдром FXTAS может быть охарактеризован как представитель совершенно нового класса нейродегенераций, имеющий свою собственную молекулярную основу (Hagerman P.J., Hagerman R.J.).
С учетом того факта, что какие-либо даже минимальные проявления синдрома FXTAS не характерны для носителей более тяжелой мутации гена FMR1 (т.е. для больных с умственной отсталостью типа FRAXA), можно заключить, что именно «промежуточный» (но не максимально длинный!) размер CGG-экспансии ответственен за патогенез FXTAS.
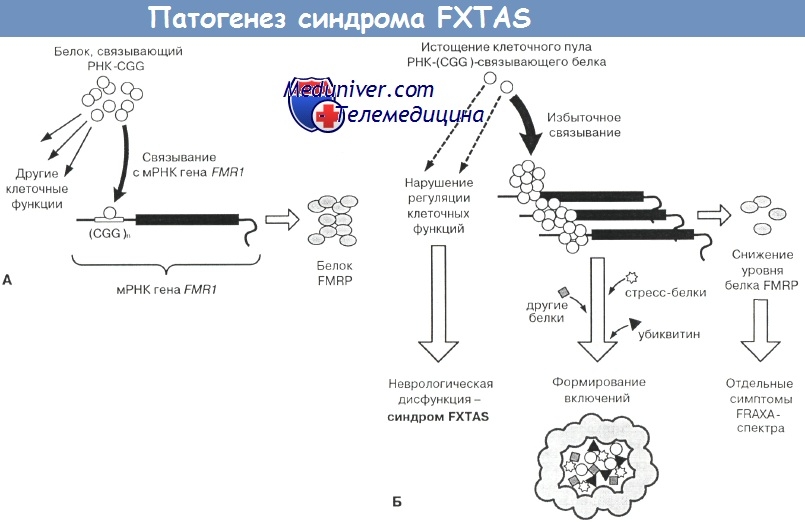
При этом следует подчеркнуть, что не имеется простой «механической» связи между тяжестью проявлений и дефицитом соответствующего белка FMRP в тканях: у больных с тяжелым синдромом FXTAS уровень белка лишь незначительно снижен, тогда как при умственной отсталости FRAXA данный белок в тканях практически отсутствует (Hagerman P.J., Hagerman R.J.). Ключом к пониманию молекулярной сущности мутации явилось обнаружение у больных FXTAS повышенного содержания мутантной мРНК гена FMR1 в тканях, по-видимому, вследствие нарушения трансляции мРНК с удлиненной тринуклеотидной цепью (Greco C.М. et al., Jacquermont S. et al.). Повышение уровня мутантной мРНК у больных FXTAS позволило предложить для объяснения патогенеза данного синдрома модель токсической функции РНК (Hagerman P.J., Hagerman R.J.). Эта модель впервые была обоснована для миотонической дистрофии — наследственного тринуклеотидного заболевания с аналогичными молекулярными характеристиками (Ranum L.P., Day J.W.).
Сущность токсической РНК-модели схематично представлена на рисунке 23 (Hagerman P.J., Hagerman R.J.). Удлиненный участок транскрипта FMR1 (имеющий увеличенное число копий CGG и измененную конформацию в составе избыточного числа молекул мРНК) обладает повышенным сродством к CGG-связывающим белкам. В результате этого:
а) экспоненциально растущий РНК-белковый комплекс становится «ядром» полимеризации в клетке, приводя к формированию включений и блокаде защитной убиквитин-протеасомной системы;
б) рекрутирование CGG-связывающих белковых молекул в состав включений приводит к истощению клеточного пула данных белков и нарушению их естественных регуляторных функций в клетке.
При умственной отсталости типа FRAXA такого не происходит, поскольку гиперэкспансия CGG-триплетов сопровождается тотальным ингибированием транскрипции гена FMR1 и практически полным отсутствием соответствующей мРНК в клетке. Подтверждением вышеуказанной «РНК-токсической» модели патогенеза синдрома FXTAS являются результаты экспериментов с трансгенными животными: при экспрессии в геноме мыши и дрозофилы CGG-фрагмента с «промежуточным» числом копий повторов (90-100) у таких трансгенных организмов формировались типичные убиквитин-позитивные включения и дегенеративные изменения в нейронах мозга и сетчатки глаза (Jin P. et al., Willemsen R. et al.).
«РНК-токсическая» модель патогенеза нейродегенерации - принципиально новый феномен в генетике человека и нейрогенетике. Его дальнейшая разработка чрезвычайно перспективна как для понимания молекулярных основ функционирования нервной системы, так и для внедрения новых патогенетических (в том числе генно-инженерных) методов лечения нейродегенеративных заболеваний. В настоящее время, помимо синдрома FXTAS и миотонической дистрофии, к заболеваниям с таким патогенезом предположительно относят также 3 формы аутосомно-доминантных атаксий с гиперэкспансией повторов в некодирующих областях - СЦА8, СЦА10 и СЦА12.
а — нормальный аллель гена FMR1; б - «премутантный» аллель гена FMR1.
- Читать "Диагностика синдрома FXTAS. ДНК-анализ"
Оглавление темы "Наследственные Х-сцепленные рецессивные атаксии":- Спиноцеребеллярная атаксия с аномальными саккадами — синдром SCASI
- Х-сцепленные рецессивные атаксии - история изучения, классификация
- Синдром FXTAS - причины, генетика
- Клиника и патогенез синдрома FXTAS
- Диагностика синдрома FXTAS. ДНК-анализ
- Лечение и профилактика синдрома FXTAS. Пренатальная диагностика
- Фридрейхоподобная Х-сцепленная атаксия - причины, клиника, диагностика
- Фатальная Х-сцепленная атаксия с глухотой и слепотой — синдром Артса
- X-сцепленные формы спастической атаксии - причины, клиника, диагностика
- X-сцепленная атаксия с поздней деменцией - причины, клиника, диагностика