
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Оливопонтоцеребеллярная атрофия (ОПЦА) - клиника, диагностика
В своем классическом описании оливопонтоцеребеллярная атрофия (ОПЦА) представляет собой медленно прогрессирующее заболевание, которое манифестирует обычно после 40—50 лет и проявляется нарастающей мозжечковой атаксией в сочетании с дизартрией, дисфонией, нарушением глотания, паркинсонизмом, пирамидными знаками, расстройством сфинктеров, реже — снижением когнитивных функций, нарушением следящих движений глазных яблок, снижением вибрационной чувствительности.
Синдром паркинсонизма занимает особое место в ряду дополнительных симптомов ОПЦА, будучи важным ключом к адекватной диагностике болезни. По мнению R. Duvoisin, около 5—6% всех пациентов с паркинсонизмом, впервые направляемых к неврологу, могут иметь ОПЦА. С другой стороны, более чем у половины больных ОПЦА при детальном неврологическом осмотре выявляются те или иные проявления паркинсоновского синдрома (брадикинезия, гипомимия, трудности в инициации ходьбы, гораздо реже —тремор покоя) (Eadie M.J., Berciano J.).
Своеобразное сочетание мозжечковой атаксии и паркинсонизма приводит к весьма необычному, «комбинированному» типу расстройства походки; иногда вследствие брадикинезии, мышечной ригидности и характерных для паркинсонизма постуральных феноменов («примерзание» в процессе ходьбы, нарушение вертикальной позы, падения) собственно мозжечковая атаксия может в какой-то степени «маскироваться» паркинсоновским симптомокомплсксом.
Именно закономерное сочетание мозжечковой атаксии с паркинсонизмом и другими многообразными симптомами при ОПЦА привело к постепенному пересмотру взглядов на ее нозологическую принадлежность. Исследователями было отмечено, что сходная с ОПЦА симптоматика и нейроморфология имеет место еще при двух редких нейродегенеративных заболеваниях — стриатонигральной дегенерации и синдроме Шая—Дрейджера.
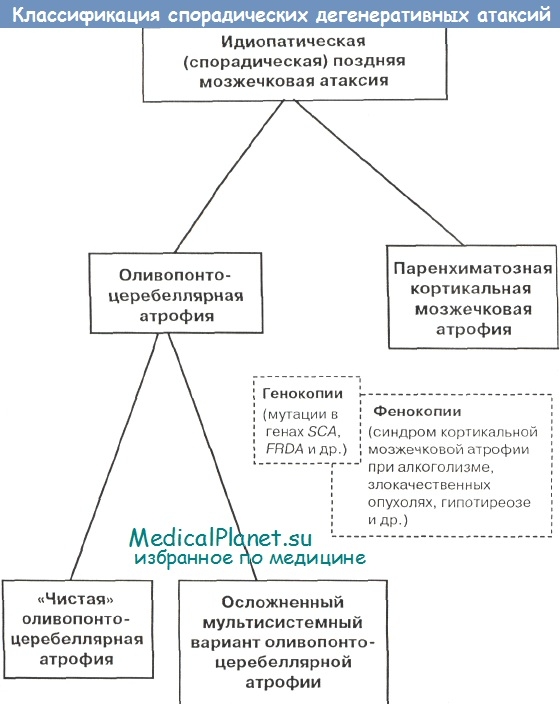
Это позволило объединить оливопонтоцеребеллярной атрофии (ОПЦА), стриатонигральную дегенерацию и синдром Шая-Дрейджера в рамках так называемой множественной системной атрофии (термин введен в практику J.Graham и D.Oppcnhcimer в 1969 г.). Концепция множественной системной атрофии как особой формы нейродегенерации предполагает возможность развития у разных пациентов вариабельного сочетания мозжечковых/пирамидных симптомов, стриарного паркинсонизма и вегетативных расстройств различной степени выраженности, а также гибель нейронов, глиоз и накопление характерных цитоплазматических включений в олигодендроглиоцитах пораженных областей мозга (Iizuka R., Wenning G. et al., Penney J.).
Для всех указанных фенотипических вариантов множественной системной атрофии характерен поздний возраст начала болезни (обычно на 5-6-м десятилетии жизни) и неуклонное прогрессирование процесса, приводящее к гибели в среднем через 6—10 лет от момента появления первых симптомов (Iizuka R., Quinn N., Wenning G. et al.).
Оливопонтоцеребеллярный вариант болезни, как указывалось выше, проявляется шаткостью походки, нарушением координации движений, расстройством речи по типу дизартрии и другими бульбарными симптомами, к которым в различных сочетаниях могут присоединяться проявления паркинсонизма и прогрессирующей вегетативной недостаточности (ортостатическая гипотензия, недержание мочи, импотенция). При стриатонигральной дегенерации, при том же наборе симптомов, «ядром» клинической картины является стриарный паркинсонизм, нередко в сочетании с негрубыми мозжечковыми, пирамидными и/или вегетативными нарушениями.
Наконец, в случае развития варианта множественной системной атрофии типа синдрома Шая—Дрейджера доминирующим клиническим признаком уже в самой ранней стадии болезни является ортостатическая гипотензия и другие вегетативные нарушения, проявляющиеся «в обрамлении» других типичных симптомов множественной системной атрофии.
Возможная общность всех трех клинических вариантов множественной системной атрофии подтверждается не только «перекрестным» характером их клинических проявлений, но и данными ПЭТ-исследований с различными лигандами, показавших в целом достаточно близкий паттерн метаболических нарушений в мозжечке, стриатонигральной системе, таламусе и коре у больных с различными фенотипами множественной системной атрофии (Penney J., Rinne J. et al.).
Наблюдавшиеся различия ПЭТ-картины ОПЦА и других вариантов множественной системной атрофии носили скорее количественный, нежели качественный характер (соотношение выраженности изменений в мозжечково-стволовой и стриарной областях мозга). Тем не менее, по мнению S.Gilman с соавт., говорить по данным ПЭТ о полной идентичности метаболических изменений в мозге при ОПЦА и в общей группе множественной системной атрофии пока преждевременно (Gilman S., Quinn N.P., Gilman S. et al.).
Вопрос о взаимосвязи ОПЦА и множественной системной атрофии был детально изучен в недавней работе американских исследователей, изучавших на протяжении 14 лет клиническую эволюцию большой серии случаев «чистой» спорадической ОПЦА (Gilman S. et al.). Авторы показали, что через 5 лет от начала болезни у 25% больных имелась трансформация ОПЦА в развернутую множественную системную атрофию — т.е. к симптомам мозжечково-стволовой дисфункции присоединились проявления паркинсонизма, вегетативной недостаточности, вовлечения кортико-спинального тракта и др.
За весь период наблюдения уже свыше 1/3 наблюдавшихся случаев «чистой» ОПЦА трансформировались в множественную системную атрофию, и эта цифра, по-видимому, занижена, поскольку часть пациентов более позднего возраста (т.е. имевших наибольший риск такой трансформации) умерли в процессе многолетнего наблюдения.
Полученные результаты могут свидетельствовать о том, что ОПЦА является гетерогенным синдромом. В части случаев у пациентов на фоне прогрессирования процесса картина болезни ограничивается поражением мозжечка, его связей и структур различных уровней ствола мозга; в этих случаях «чистой» ОПЦА болезнь протекает относительно благоприятно и может растянуться на годы и даже десятилетия (Gilman S. et al.).
В других случаях оливопонтоцеребеллярной атрофии (ОПЦА) с течением времени закономерно осложняется присоединением совокупности симптомов поражения других областей головного и спинного мозга и эволюционирует, таким образом, в типичную картину множественной системной атрофии; этот вариант течения ОПЦА является гораздо более злокачественным и заканчивается фатальным исходом в среднем через 7 лет от начала болезни (Klockgcther T. et al., Gilman S. et al.). Именно такое современное деление ОПЦА на «чистую» и мультисистемную формы представлено на классификационной схеме на рисунке.
В связи с гетерогенностью оливопонтоцеребеллярной атрофии (ОПЦА) весьма важным с практической точки зрения является вопрос об определении прогноза у конкретных пациентов с впервые диагностированным заболеванием. По имеющимся данным, о высоком риске трансформации ОПЦА во множественную системную атрофию может свидетельствовать поздний возраст начала атаксии (старше 51 года), быстрый темп нарастания расстройств координации в дебюте болезни, а также раннее развитие дисфункции со стороны мочеполовой сферы (Wenning G.K., Gilman S. et al.). Более позднее начало болезни повышает риск неблагоприятного течения процесса и быстрой гибели больных (Ben-Shlomo Y. et al., Klockgcther T. et al.).
- Читать "Морфология оливопонтоцеребеллярной атрофии (ОПЦА)"
Оглавление темы "Идиопатические атаксии":- Атаксия при метаболических лейкодистрофиях - клиника, диагностика
- Атаксия при врожденных болезнях гликозилирования - клиника, диагностика
- Идиопатическая поздняя мозжечковая атаксия - определение, история изучения
- Классификация идиопатической поздней мозжечковой атаксии
- Оливопонтоцеребеллярная атрофия (ОПЦА) - клиника, диагностика
- Морфология оливопонтоцеребеллярной атрофии (ОПЦА)
- Паренхиматозная кортикальная мозжечковая атрофия - клиника, диагностика
- Диагностика идиопатической поздней мозжечковой атаксии
- Дифференциальная диагностика идиопатической поздней мозжечковой атаксии
- Пример идиопатической поздней мозжечковой атаксии