
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Роль патологии митохондрий в патогенезе атаксии Фридрейха
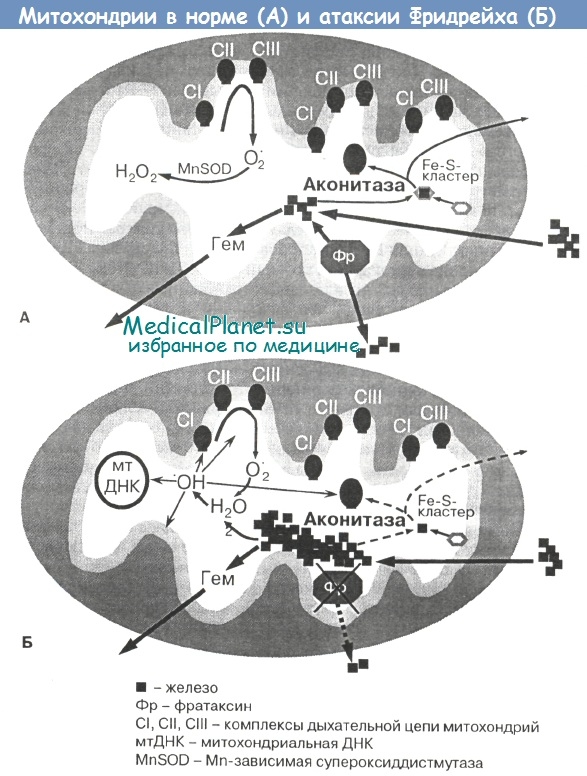
Митохондрии, вероятно, играют основную роль в регуляции распределения железа в клетке (то же самое справедливо и в отношении кальция). При атаксии Фридрейха фратаксин, количество и функциональная активность которого в мембране митохондрий снижены, не может качественно реализовать свою «поглотительную» функцию в отношении избыточного железа. Недостаточность фратаксина приводит к аккумуляции железа цитозольного происхождения внутри митохондрий (Pandolfo М.).
С повышением содержания железа в митохондриях более чем в 10 раз общее клеточное железо остается в пределах нормальных значений, а содержание цитозольного железа снижается. Это приводит к патологической стимуляции (10—50-кратное повышение активности) высокоаффинной к железу транспортной системы, содержащей ряд ферментов (таких как ферроксидаза и пермеаза, которые обычно не экспрсссируются в насыщенной железом среде), что сопровождается развитием системного дефекта метаболизма железа в клетке.
В экспериментах показано, что высокоаффинные железотранспортные системы в нормальных дрожжевых клетках активизируются только при росте в питательной среде с пониженным содержанием железа, а дрожжевые клетки с «нокаутированным» гомологом фратаксина проявляют высокую активность железотранспортной системы даже в среде с избыточным содержанием этого иона (Wilson R. et al.).
Первые прямые доказательства сходства механизмов развития патологического процесса у дрожжей с дефектом фратаксина и в клетках человека при атаксии Фридрейха были получены M. Delatycki и соавт в процессе изучения культур фибробластов, полученных от больных атаксией Фридрейха: уровень железа в митохондриальной фракции у больных был значительно выше, чем у здоровых индивидуумов.
Авторами не было обнаружено снижения митохондриальной ДНК относительно общей геномной ДНК в фибробластах, аналогично результатам исследований этого показателя в лимфобластах, кардиомиоцитах и нейронах (Campuzano V. et al.). В целом, основываясь на многочисленных экспериментальных исследованиях, можно заключить, что патофизиологические процессы в мутантных дрожжевых клетках и клетках тканей пациентов с атаксией Фридрейха чрезвычайно схожи.
У всех позвоночных первичным регулятором клеточного метаболизма железа является мРНК-связанный белок IRE-BPI. Он образуется при отщеплении Fe-S-кластера от цитозольной аконитазы. Когда железа в цитозоле клетки недостаточно, IRE-BP1 связывает 3'-конец мРНК трансферринового рецептора, защищая его тем самым от деградации и позволяя клетке вводить большее количество железа.
Когда цитозольное железо содержится в клетке с избытком, IRE-BP1 соединяется с Fe-S-кластеросодержащим белком, тем самым стимулируя трансляцию мРНК ферритина и уменьшая количество мРНК трансферриновых рецепторов. По мере расходования цитозольного железа синтез ферритина уменьшается. Таким образом, с современных позиций атаксия Фридрейха рассматривается как состояние с аномальным распределением клеточного железа (Pandolfo M.).
В подтверждение данной концепции опубликован ряд работ, в которых авторами было показано:
1) наличие зон дегенерации сердечной мышцы с множественными депозитами железа внутри миокардиальных волокон, выявленное при атаксии Фридрейха post mortem (Lamarche J. et al.);
2) избыточное содержание железа в кардиомиоцитах (а иногда в печени и селезенке) на биопсийном материале, полученном от больных с атаксией Фридрейха (Rustin P. et al., Knight S. et al., Bradley J. et al.);
3) косвенное подтверждение повышения уровня железа в nucleus dentatus, выявленное при MPT в режиме мультиградиент-эхо (Waldvogel D. et al.);
4) повышенная чувствительность культуры лимфобластов, полученных от пациентов с атаксией Фридрейха, к солям ионов металлов с переменной валентностью.
При этом в экспериментах in vitro показано наличие избирательной повышенной чувствительности клеток только к FeCl3 и MnСl2, но не к другим металлам в составе солей (СоCl2, CuSO4, ZnCl2 и др.) (Wong A. et al.).
Биохимические процессы в системе окислительного фосфорилирования и дыхательной цепи митохондрий являются мощным источником свободнорадикальных форм кислорода в клетке. При этом металлы переменной валентности, в том числе железо, играют важную роль в катализе реакций с образованием активных форм кислорода с высоким редокс-потенциалом. При различных патологических состояниях эти реакции могут усиливаться.
В норме постоянное образование прооксидантов в клетке уравновешено той же скоростью их дезактивации антиоксидантными системами. Резкое усиление окислительных процессов при недостаточности или истощении системы антиоксидантной защиты приводит к развитию окислительного стресса, который в настоящее время рассматривается как один из общих механизмов повреждения тканей организма. Известно, что одним из наиболее токсичных интермедиатов кислорода, имеющих самый высокий редокс-потенциал (т.е. наиболее высокую реакционную способность к окислению), является гидроксильный радикал ОН. Образование ОН в биологических системах происходит в ходе реакции Фентона с участием металлов переменной валентности, главным образом Fe2+, из менее токсичных активных форм кислорода (перекиси водорода и супероксидного анион-радикала):
Н2О2 + Fe (II) -> Fe (III) + ОН- + ОН-
Гидроксильный радикал ОН чрезвычайно активно взаимодействует с липидами, образуя липидные гидроперекиси в реакциях перекисного окисления липидов (ПОЛ). Таким образом, аккумуляция железа внутри митохондрий при атаксии Фридрейха увеличивает продукцию свободных радикалов в ходе фентоновской реакции, и митохондрии утрачивают способность эффективно осуществлять окислительное фосфорилирование (Pandolfo М., Foury F, Schulz J. et al., ShererT. et al.). Следствием этого является снижение синтеза АТФ, а также дисфункция и необратимое повреждение клетки в условиях системного энергетического дефицита.
R.Lodi и J.Cooper в 1999 г., используя метод магнитно-резонансной спектроскопии, показали in vivo трехкратное снижение скорости синтеза АТФ в мышечной ткани больных атаксией Фридрейха по сравнению со здоровыми испытуемыми в условиях покоя, физической нагрузки и восстановления после нагрузки, причем выявленное снижение окислительного фосфорилирования коррелировало с числом GAA-повторов в гене FRDA. Системные нарушения функции митохондрий при атаксии Фридрейха были показаны также в работе M.Vorgerd и соавт..
Оценка окислительного стресса при атаксии Фридрейха in vivo была осуществлена M.Emond и соавт. при анализе уровня малонового диальдегида — продукта ПОЛ — в плазме крови у 11 больных методом высокоэффективной жидкостной хроматографии: содержание малонового диальдегида оказалось в Образа выше, чем у здоровых индивидуумов (Emond М. et al.). При этом значимой корреляции содержания малонового диальдегида с продолжительностью заболевания или числом GAA-повторов авторами не было выявлено. M.Shigenaga и соавт. обнаружили в моче больных атаксией Фридрейха 3-кратное повышение концентрации 8-гидрокси-2-d-оксигуанозина (8ОH2'dG) - чувствительного биологического маркера окислительного повреждения ДНК in vivo (Shigenaga М. et al.).
Примечательно, что авторы обнаружили относительную специфичность данного показателя, поскольку при других неврологических заболеваниях, таких как миопатии, деменции, рассеянный склероз, периферические невропатии и мультисистемная атрофия, уровень 80H2'dG оставался нормальным; похожее повышение содержания оксигуанозина наблюдалось лишь при боковом амиотрофическом склерозе, при котором образование активных форм кислорода играет важную роль в патогенезе заболевания (Bogdanov M.et al.).
В реализации дальнейшего патогенетического каскада несомненное значение имеет избыточный вход кальция в клетку, инициирующий реакции апоптоза. На модели фибробластов больных атаксией Фридрейха показано, что добавление в питательную среду хелатного соединения, удаляющего кальций из клетки, защищает культуру фибробластов от гибели (Wong A. et al.). Подобный эффект наблюдался также при культивировании фибробластов в среде, обедненной ионами кальция. Значимость апоптоза при атаксии Фридрейха подтверждается значительной активацией у больных проапоптотического фермента каспазы-9 и выявлением маркеров гиперактивности «сигнальных» стрессовых путей клетки (Pianesc L. et al.).
- Читать "Роль нарушений обмена аминокислот в патогенезе атаксии Фридрейха"
Оглавление темы "Атаксия Фридрейха":- Лечение и профилактика врожденных мозжечковых атаксий
- Атаксия Фридрейха - история изучения
- Генетика атаксии Фридрейха
- Клинико-генетические корреляции при атаксии Фридрейха
- Фратаксин - функции, значение. Патогенез атаксии Фридрейха
- Роль патологии митохондрий в патогенезе атаксии Фридрейха
- Роль нарушений обмена аминокислот в патогенезе атаксии Фридрейха
- Диагностика атаксии Фридрейха путем оценки митохондриальных нарушений
- Цитохимический анализ активности митохондриальных ферментов при атаксии Фридрейха
- Морфометрический анализ активности митохондриальных ферментов при атаксии Фридрейха