
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Болезни дыхательной цепи митохондрий (БДЦМ) у новорожденных - причины, диагностика, лечение
Митохондрия — это округлая или овальная структура; органоид клетки, состоящий из двуслойной мембраны, гладкая сторона которой обращена кнаружи, а внутренняя представляет собой складчатую структуру, образующую так называемые кристы. Митохондрия является основным источником энергии в клетке, содержащим цитохромы, ферменты цикла лимонной кислоты, окисления жирных кислот и окислительного фосфорилирования. Дыхательные (респираторные) комплексы располагаются на внутренней мембране митохондрий.
В данной статье описана митохондриальная патология, приводящая к поражению нервной системы у новорожденных и детей первых лет жизни, а также представительная группа нарушений b-окисления митохондриальных жирных кислот (в неонатальном периоде).
Болезни дыхательной цепи митохондрий (БДЦМ) — большая, представительная и гетерогенная группа различных наследственных болезней обмена веществ, обусловленных нарушениями работы комплексов дыхательной цепи митохондрий. БДЦМ нередко называют «новым классом болезней»; к настоящему времени их насчитывается около 40.
Еще в 1962 г. R. Luft и соавт. впервые обратили внимание на взаимосвязь мышечной слабости с нарушениями процессов окислительного фосфорилирования в митохондриях мышечной ткани. RE Chinnery и соавт. указывают, что патологические мутации ДНК митохондрий обнаруживаются у 1 из 8000 индивидов.
Болезни дыхательной цепи митохондрий (БДЦМ) (митохондриальные энцефаломиопатии) — это наследственная патология, возникающая в результате нарушений клеточной энергетики, характеризующаяся полиморфизмом клинических проявлений, выражающаяся в поражении преимущественно ЦНС и мышечной системы, а также других органов и систем организма.
Болезни дыхательной цепи митохондрий (БДЦМ) характеризуются изолированным или сочетанным дефицитом комплексов дыхательной цепи митохондрий.
Разновидности митохондриальной патологии в неврологии сравнительно многочисленны, они включают следующие патологические состояния (три основные категории): митохондриальные синдромы с «рваными красными волокнами» (синдром MELAS — митохондриальная энцефалопатия с лактат-ацидозом и инсультоподобными эпизодами), синдром MERFF (миоклоническая эпилепсия с рваными красными волокнами), синдром Кирнса-Сейра, прогрессирующая наружная офтальмоплегия/РЕО, синдром Пирсона, синдром множественных делеций митохондриальной ДНК; митохондриальные синдромы без «рваных красных волокон» (энцефалопатия Лея/подострая перивентрикулярная некротизирующая энцефалопатия, NARP/нейропатия, атаксия, пигментный ретинит, синдром деплеции митохондрий раннего младенческого возраста, наследственная оптическая нейропатия Лебера/LHON, синдром Вольфрама, синдром Барта; дефекты митохондриального транспорта/миопатии липидного хранения (недостаточность карнитин-пальмитоилтрансферазы, липидная миопатия с нормальными уровнями карнитина и др).
В зависимости от наличия основного метаболического дефекта рассматривают четыре основных группы болезней дыхательной цепи митохондрий (БДЦМ):
1) нарушения обмена пирувата;
2) дефекты обмена жирных кислот (ЖК);
3) нарушения цикла Кребса;
4) дефекты электронного транспорта и окислительного фосфорилирования (OXPHOS).
Причиной их возникновения являются мутации в генах, кодирующих белки, задействованные в процессах энергоообмена в клетках, включая субъединицы комплекса пируватдегидрогеназы, ферменты цикла Кребса, компоненты цепи транспорта электронов (ЦТЭ), структурные белки ЦТЭ, митохондриальные транспортеры внутренней мембраны, регуляторы митохондриального нуклеотидного пула, а также факторы, взаимодействующие с ДНК митохондрий (mtDNA).
В подавляющем большинстве случаев отмечается вовлечение ЦТЭ в патологический процесс (первичное или вторичное). Компоненты ЦТЭ частично кодируются ядерными генами, вследствие чего часть болезней дыхательной цепи митохондрий наследуется по аутосомно-рецессивному типу (через ДНК митохондрий).
Н.В. Sarnat и J.H. Menkes, считают, что митохондриальные нарушения у детей связаны с большим числом болезней, не являющихся первичными митохондриальными цитопатиями, но при которых нарушения функций митохондрий вносят значимый вклад в патогенез и клинические проявления заболеваний; эти болезни могут быть метаболическими, дегенеративными, воспалительными, врожденными и/или приобретенными мальформациями, а также неоплазмами.
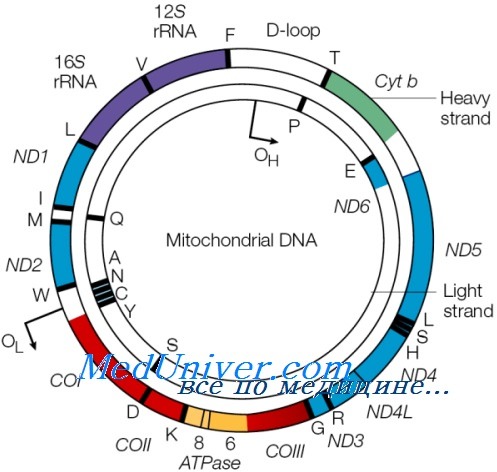
Генетические аспекты митохондриальной патологии сложны и неоднозначны. Болезни дыхательной цепи митохондрий передаются по материнскому (митохондриальному) типу наследования, а также по аутосомно-рецессивному, аутосомно-доминантному и Х-сцепленному типам (мутации в ядерном геноме). У митохондрий имеется собственная ДНК (mtDNA) на единичной округлой хромосоме (около 16 kb или 16 т.н.): kb (англ. kilobase) — тысяча нуклеотидов (т.н.); единица измерения длины молекулы нуклеиновой кислоты; для 2-цепочечных молекул т.н. соответствует тысяче пар нуклеотидов (т.п.н.).
Митохондриальная ДНК тесно взаимодействует с ДНК ядра (nDNA), а в каждом из 5 известных дыхательных комплексов основная часть субъединиц кодируется nDNA, а не mtDNA. Комплекс I состоит из 41 субъединицы, из которых 7 кодируются mtDNA; а остальные — ядерной ДНК. Комплекс II обладает всего четырьмя субъединицами; большая часть кодируется ДНК. Комплекс III представлен 10 субъединицами (кодирование mtДНК — 1, nДНК — 9). Комплекс IV имеет 13 субъединиц, из которых 3 кодируются mtДНК, а остальные 10 — пДНК (ядерная ДНК). Комплекс V включает 12 субъединиц, кодирование mtДНК — 2, nДНК — 10.
Практически всегда mtДНК наследуется по материнской линии (митохондриальный тип). Такой тип наследования очевиден в значительной части случаев передачи митохондриальных заболеваний. Остальные случаи обычно относятся к аутосомно-рецессивному типу передачи, являются спорадическим или непредсказуемыми.
Поскольку геном ядра поставляет соответствующие субъединицы дыхательным комплексам, некоторые митохондриальные болезни могут наследоваться по менделевскому типу, когда метаболический эффект обусловливается нарушениями субъединиц, кодируемых мутациями пДНК (с сохранением нормальных mtДНК). При таких мутациях наследование почти всегда оказывается аутосомно-рецессивным.
При гомоплазмии все молекулы mtДНК в пределах клетки идентичны, но при митохондриальном наследовании в клетке одновременно сосуществуют 2 популяции mtДНК (состояние гетероплазмии). Большинство патогенетически значимых мутаций mtДНК являются гетероплазмичными, когда соотношение между нормальными и аномальными mtДНК определяет клиническую выраженность и тяжесть митохондриальных заболеваний (наличие 100% аномальной mtДНК считается не совместимым с жизнью). Кроме того, гетероплазмия может отмечаться не только на клеточном уровне, но и на уровне митохондрий (интрамитохондриальная гетероплазмия).
Во время деления клеток (митоза) митохондрии и mtДНК дифференцируются на 2 дочерние клетки (митотическая сегрегация). Соотношение нормальных и аномальных mtДНК, наследуемое каждой из дочерних клеток, не обязательно одинаково, а пороговый эффект мутации mtДНК может влиять на клинические проявления, включая возраст пациента к моменту дебюта болезни.
Еще одним классом нарушений mtДНК являются так называемые «крупномасштабные перестройки mtДНК», такие как делеция или вставка т.н. (kb) митохондриальной хромосомы.
Многие точечные мутации mtДНК и ядерной ДНК пока не изучены, в частности при митохондриальных миопатиях «с рваными красными волокнами», некоторых митохондриальных энцефалопатиях (особенно при энцефалопатии Лея), а также при митохондриальных кардиомиопатиях. Их вклад в развитие клинической картины БДЦМ еще не ясен.
Митохондриальная патология исключительно вариабельна по тяжести, клиническим проявлениям и возрасту к моменту дебюта болезни. Симптоматика патологических состояний, входящих в понятие «митохондриальная патология», может простираться от приступов головных болей до летального мультисистемного (полиорганного) поражения у младенцев. Проявления болезни обычно возникают, когда местное поступление энергии оказывается неадекватным потребностям новорожденного, поэтому чаще поражаются те органы и ткани, которым требуется наибольшее количество энергии: нервная и мышечная ткани, эндокринные железы, а также почечный тубулярный эпителий. Среди состояний, относящихся к митохондриальной патологии, обычно доминируют заболевания головного мозга и мышечной системы, они чаще рассматриваются в качестве энцефаломиопатий.
Энцефаломиопатии — любое поражение ЦНС и мышечной системы; при неврологических формах митохондриальной патологии — это гетерогенная группа нарушений с вариабельными клиническим течением и симптоматикой.
Если согласиться с утверждением A. Munnich, что «митохондриальные заболевания могут вызывать любой симптом в любой ткани, в любом возрасте, при любом типе наследования», то в неонатальном периоде можно ожидать манифестации любой формы митохондриальных энцефаломиопатий.
Наиболее часто повреждаемые органы и системы (изолированно или в сочетании) при митохондриальной патологии
| Системы и органы | Анатомические структуры | Клинические проявления |
| Нервная система |
Кора.
Мозжечок. Базальные ганглии. Ствол. Сетчатка (retina). Улитка (cochlea). Периферические нервы. Автономная иннервация |
Судороги, ЗПМР.
Атаксия. Хорея, дистония. Апноэ, рвота. Ретинопатия, атрофия ДЗН. Сенсоневральная тугоухость. Периферические нейропатии. Дисавтономия, нарушения перистальтики ЖКТ |
| Мышечная система |
Большие скелетные мышцы.
Наружные глазные мышцы. Сердечная мышца. Гладкая мускулатура |
Гипотония, слабость.
Слабость мышц глаз, птоз. Кардиомиопатия, аритмия. Нарушения подвижности ЖКТ |
| Эндокринные органы |
Щитовидная железа.
Островки Лангерганса. Надпочечники. Гипофиз |
Гипотиреоз.
Сахарный диабет. Болезнь Аддисона. Недостаточность гормона роста, гипогликемия |
| Почки | Тубулярный эпителий | Ацидоз, амино- и органоацидурия Фанкони |
В таблице представлены наиболее часто повреждаемые системы и органы, требующие энергии, а также перечислены основные клинические проявления у детей с митохондриальной патологией (цитируется по Boles R. и Moseley К.).
Органы и системы, требующие энергии в умеренном количестве, нередко повреждаемые при митохондриальной патологии, обычно в сочетании с одним или большим числом вышеперечисленных проявлений, представлены в таблице ниже.
Системы и органы, требующие энергии в умеренном количестве, нередко повреждаемые при митохондриальной патологии
| Системы и органы | Анатомические структуры | Клинические проявления |
| Кроветворная система | Костный мозг | Анемия/нейтропения/тромбоцитопения |
| Экзокринные железы | Поджелудочная железа | Панкреатит |
| Почки | Нефрон | Почечная недостаточность |
У новорожденных с митохондриальными нарушениями часто отмечаются нарушения ЖКТ, абдоминальные боли, тошнота и рвота, а по завершении неонатального периода — отставание в физическом развитии (линейный рост) и ожирение. Эти симптомы могут быть вторичными по отношению к основному заболеванию. Наступление болезни наиболее вероятно при подверженности новорожденного стрессу, т.е. когда отмечаются самые высокие потребности в энергии (при голодании, физической нагрузке, инфекции и т.д.). Попытки выявить корреляции между клиническими симптомами и специфическими дефицитарными нарушениями дыхательного комплекса пока не принесли успеха. Ряд биохимических изменений (активность специфических ферментов) вторичен, а сами дефекты дыхательной цепи у новорожденных могут быть неспецифическими.
Для диагностики митохондриальной патологии, помимо клинических признаков болезней, применяются мышечная и кожная биопсия, электронная микроскопия, количественное определение комплексов дыхательной цепи и молекулярно-генетический анализ.
Определение содержания концентраций в крови лактата и пирувата (а также их соотношения) на фоне нагрузки глюкозой — ориентировочный критерий диагностики митохондриальных болезней, как и анализ содержания в крови жирных кислот и продуктов перекисного окисления, а также определение в моче содержания органических кислот.
Биохимические показатели не могут считаться четким диагностическим критерием митохондриальной патологии. Диагностически более значимым является определение в крови уровней карнитина, а в различных тканях организма — митохондриальных ферментов.
При помощи методов нейровизуализации (КТ, МРТ) у новорожденных обнаруживаются патологические изменения митохондрий нейронов, глиальных и эндотелиальных клеток, но указанные изменения не всегда патогномоничны для митохондриальной патологии (могут присутствовать при других заболеваниях ЦНС).
Секвенирование определенных участков митохондриального генома и ядерных генов позволяет проводить выявление точечных мутаций, крупных делеций и вставок в гене. Метод полимеразной цепной реакции (ПЦР) может применяться при исследовании крови для обнаружения делеций ДНК митохондрий при некоторых митохондриальных миопатиях. В настоящее время найдены частые мутации для такой патологии, как синдром Кирнса-Сейра, MELAS, MERRF, РЕО, энцефалопатия Лея, LHON и др.
Морфологическое исследование биоптата скелетных мышц позволяет с достаточной точностью выявлять «рваные красные волокна» (традиционная и электронная микроскопия), хотя оно инвазивно. Цитохимический анализ митохондриальной активности в клетках крови (лимфоциты) позволяет выявлять полисистемные митохондриальные нарушения.
Общепринятое лечение БДЦМ не разработано, если не считать попыток применения коэнзима Q , реамберина и креатина, среди которых коэнзим Q и креатин можно рассматривать в составе диетотерапии как кофактор диеты.
В периоде новорожденности у детей наиболее часто встречается синдром (энцефалопатия) Лея.
- Вернуться в оглавление раздела "неврология"
Оглавление темы "Неврология болезней обмена веществ":- Гипервалинемия у новорожденных - причины, диагностика, лечение
- Гомоцистинурия у новорожденных - причины, диагностика, лечение
- Метилмалоновая ацидемия (ММА) у новорожденных - причины, диагностика, лечение
- Пропионовая ацидемия (ППА) у новорожденных - причины, диагностика, лечение
- Глутаровая ацидемия тип I (ГАI) у новорожденных - причины, диагностика, лечение
- Кетоадипиновая ацидемия (2-КАА) у новорожденных - причины, диагностика, лечение
- Изовалериановая ацидемия (ИВА) у новорожденных - причины, диагностика, лечение
- 3-метилкротнил глицинурия (3-МКГ) у новорожденных - причины, диагностика, лечение
- Нарушения цикла метаболизма мочевины (НЦММ) у новорожденных - причины, диагностика, лечение
- Болезни дыхательной цепи митохондрий (БДЦМ) у новорожденных - причины, диагностика, лечение