
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Классификация идиопатической поздней мозжечковой атаксии
Несмотря на очевидные различия развернутой морфологической картины оливопонтоцеребеллярной атрофии и паренхиматозной кортикальной мозжечковой атрофии рядом авторов справедливо отмечалась сложность клинического подразделения идиопатической спорадической мозжечковой атаксии на 2 указанных подтипа, особенно в ранней стадии болезни.
Путаницу добавляет имеющееся у части пациентов с оливопонтоцеребеллярной атрофией и кортикальной атрофией дополнительное вовлечение в дегенеративный процесс спинного мозга или базальных ганглиев. В связи с этим в литературе неоднократно предпринимались попытки разработать более приемлемую клиническую классификацию спорадических форм дегенеративных мозжечковых атаксий. Наиболее известной является классификация A. Harding, согласно которой данные заболевания могут быть разделены на 3 основных подтипа (Harding А.Е.):
• подтип А - характеризуется относительной сохранностью координации движений в конечностях (особенно в руках) при наличии значительной туловищной атаксии при ходьбе. Дополнительными особенностями данного подтипа, по A.Harding, служат поздний возраст появления первых симптомов болезни (после 50 лет), а также нередко наблюдающиеся признаки вовлечения сенсорных путей и выпадение ахилловых рефлексов;
• подтип В — характеризуется сравнительно поздним началом и развитием выраженного постурального тремора и/или тремора покоя в конечностях. При этом интенционный тремор также является «непропорционально тяжелым» по отношению к выраженности дисдиадохокинсза. Еще в 1962 г. M.Critchley очень точно и образно определил обсуждаемый редкий фенотип как «гротескный пример эссенциального тремора». Похожие случаи были описаны ранее в качестве «прогрессирующей мозжечковой диссинергии Ханта» (Hunt J.R.), хотя ныне очевидно, что форма Ханта представляет собой сборную группу и не может считаться нозологически самостоятельной;
• подтип С — наблюдается несколько чаще и мужчин и характеризуется мультисистемностью поражения. В клинической картине имеется сочетание статико-локомоторной и динамической атаксии с различной комбинацией таких симптомов, как дизартрия, пирамидные знаки, сенсорный дефицит, гипорефлексия, экстрапирамидные расстройства, нарушение функции тазовых органов, офтальмоплегия, деменция и др.
Классификация A.Harding сыграла заметную роль в уточнении фенотипического спектра идиопатических спорадических мозжечковых атаксий, однако практика показала определенную нечеткость и условность критериев подразделения данных заболеваний на отдельные подтипы, что справедливо отмечалось рядом исследователей (Kumar D., Timperley W.R.).
Более того, внимательный анализ показывает сходство предложенных A. Harding подтипов атаксий с классическими клинико-морфологическими формами мозжечковых дегенерации: так, подтип А весьма близок к форме Мари—Фуа—Алажуанина, подтип С почти полностью соответствует описанию оливопонтоцеребеллярной атрофии Дежсрина—Тома, а подтип В представляет собой большую редкость и, по-видимому, является гетерогенным синдромом.
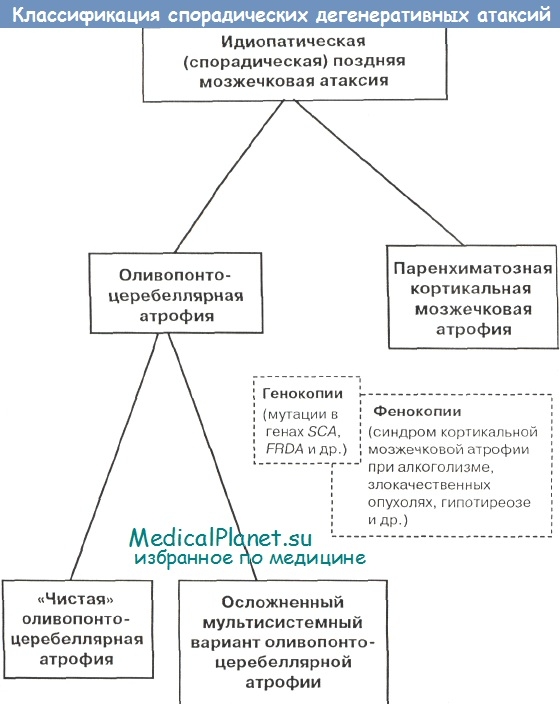
Таким образом, на сегодняшний день наиболее общепринятым остается деление идиопатической спорадической мозжечковой атаксии на оливопонтоцеребеллярную атрофию и паренхиматозную кортикальную мозжечковую атрофию. Весьма возможно, что такое деление условно и данные формы входят в спектр патогенетически и нозологически единого нейродегенеративного процесса. Современные представления о структуре идиопатической спорадической мозжечковой атаксии показаны на рисунке.
Следует помнить, что в части спорадических случаев может иметь место не «истинная» идиопатическая поздняя мозжечковая атаксия, а какой-либо известный генетический вариант наследственных атаксий (например при отсутствии достоверных сведений о семейном анамнезе).
С другой стороны, известен и ряд фенокопий идиопатических мозжечковых атаксий, которые будут подробнее рассмотрены далее (синдром кортикальной мозжечковой атаксии при алкоголизме, гипотиреозе, злокачественных новообразованиях и т.д.).
Не вызывает сомнений, что за счет указанных гено- и фенокопии в любой обследованной выборке больных доля «истинной» идиопатической мозжечковой атаксии будет ниже общего числа выявленных случаев спорадических мозжечковых атаксий позднего возраста. В связи с этим весьма затрудненной является точная оценка популяционной распространенности идиопатической поздней мозжечковой атаксии.
По имеющимся данным, она может достигать 5—10 случаев на 100 000 населения или даже составлять до 90% всех случаев атаксий дегенеративной природы (т.е. лишь 10% дегенеративных атаксий имеют наследственный характер) (Duvoisin R.G., Hagerman P.J., Hagerman R.J.). Эти оценки совпадают с эмпирическими данными о соотношении наследственных и спорадических форм атаксий среди больных, госпитализируемых в соответствующие специализированные отделения.
Так, наша собственная практика показала, что в невыборочной серии случаев дегенеративных атаксий, ежегодно проходящих обследование в нейрогснетическом отделении Института неврологии РАМН, свыше 60% больных имеют те или иные варианты идиопатической поздней мозжечковой атаксии и отрицательный семейный анамнез.
- Читать "Оливопонтоцеребеллярная атрофия (ОПЦА) - клиника, диагностика"
Оглавление темы "Идиопатические атаксии":- Атаксия при метаболических лейкодистрофиях - клиника, диагностика
- Атаксия при врожденных болезнях гликозилирования - клиника, диагностика
- Идиопатическая поздняя мозжечковая атаксия - определение, история изучения
- Классификация идиопатической поздней мозжечковой атаксии
- Оливопонтоцеребеллярная атрофия (ОПЦА) - клиника, диагностика
- Морфология оливопонтоцеребеллярной атрофии (ОПЦА)
- Паренхиматозная кортикальная мозжечковая атрофия - клиника, диагностика
- Диагностика идиопатической поздней мозжечковой атаксии
- Дифференциальная диагностика идиопатической поздней мозжечковой атаксии
- Пример идиопатической поздней мозжечковой атаксии