
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Генетика аутосомно-доминантных атаксий
По состоянию на начало 2006 г. установлено существование 27 хромосомных локусов прогрессирующих аутосомно-доминантных спиноцеребеллярных атаксий, а для 14 форм этих заболеваний идентифицированы мутантные гены и их белковые продукты (ОМ1М). В тех случаях, когда функция соответствующего белка неизвестна, гены получили обозначение ATXN1, ATXN2, ATXN3 и т.д. (от англ. «ATAXIN», производное от «ataxia»).
Необходимо добавить, что в некоторых семьях с аутосомно-доминантными атаксиями было показано достоверное исключение сцепления с известными хромосомными локусами (Giunti Р. et al., Worth P. et al.), что свидетельствует о дальнейшей генетической гетерогенности данной группы. В рамках принятой номенклатуры доминантных атаксий остались свободными рубрики «СЦА9» и «СЦА24», которые зарезервированы для пока не установленных форм атаксий.
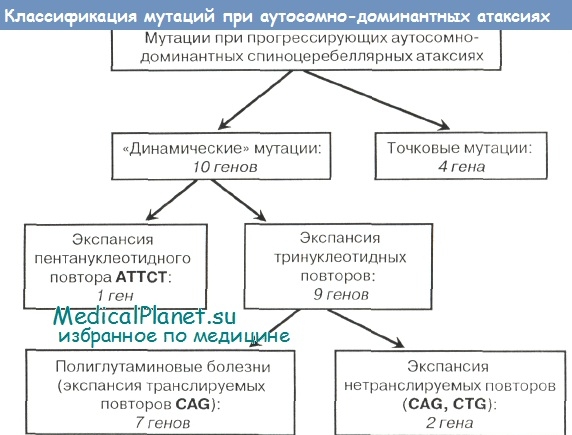
Как видно из классификации аутосомно-доминантных атаксий, при 10 из 14 форм аутосомно-доминантных спиноцеребеллярных атаксий с идентифицированными генами имеет место особый характер повреждения гена — так называемые «динамические» мутации, или мутации по типу экспансии внутривенных простых тандемных повторов. Данному классу мутаций, открытому в начале 1990-х годов и характерному для ряда нейрогередитарных заболеваний, посвящена обширная литература (Иллариошкин С.Н. и др., Brice А., Stevanin G. et al., Willems P.).
Он связан с существованием в геноме участков, представленных цепочкой одних и тех же многократно повторяющихся элементарных нуклеотидных последовательностей. Каждый такой повторяющийся элемент может состоять из 2, 3, 4 или 5 нуклеотидов — соответственно ди-, три-, тетра- или пентануклеотидные повторы. Число повторов в норме варьирует в строго определенных пределах, тогда как у больных имеется патологическое увеличение числа копий (экспансия) таких повторов, превышающее определенный порог.
Термин «динамические» применяется по отношению к рассматриваемым мутациям в связи с тем, что патологически удлиненный участок гена является нестабильным и нередко меняет свою конфигурацию при передаче гена в следующее поколение (т.е. в мейозе число триплетов может нарастать или, реже, сокращаться вплоть до нормы) (Richards R., Sutherland С., Willems P.).
На рисунке представлена более детальная молекулярная субклассификация аутосомно-доминантных спиноцеребеллярных атаксий, для которых установлены первичные генетические дефекты. Как видно на рисунке, при одной из форм атаксий — СЦА10 — выявлена экспансия пентануклеотидного повтора (АТТСТ)n в некодирующей области гена ATXN10. При остальных 9 формах атаксий, характеризующихся «динамическими» мутациями, имеет место экспансия более простых, тринуклеотидных повторов различной конфигурации.
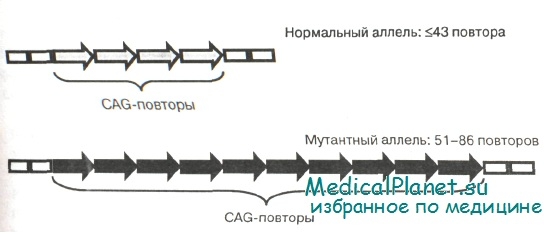
Характеристики нормального и мутантного аллелей показаны для гена ATXN3, содержащего (CAG)n-повтор в кодирующей области
Группа тринуклеотидных атаксий также неоднородна и может быть подразделена на две самостоятельных категории:
1) При формах СЦА8 и СЦА12 имеется удлинение нетранслируемых повторов CAG/CTG в соответствующих генах. Таким образом, эти мутации не являются кодирующими и не меняют последовательность аминокислот в белке (предполагается, что они могут действовать на уровне мРНК).
2) Для остальных 7 форм аутосомно-доминантных атаксий с тринуклеотидными «динамическими» мутациями характерна экспансия тандемных CAG-триплстов в кодирующих областях генов.
Поскольку триплет CAG (цитозин—аденин-гуанин) кодирует аминокислоту глутамин, на белковом уровне экспансия (CAG)n-повтора приводит к пропорциональному удлинению полиглутаминового участка в составе соответствующего белка. Таким образом, большинство молекулярных форм аутосомно-доминантных спиноцеребеллярных атаксий относятся к особому классу полиглутаминовых болезней (Иллариошкин С.Н., Ross С., Stevanine G. et al.). Патогенез данного класса нейродегенеративных заболеваний, к которым относятся также хорея Гентингтона и спинально-бульбарная амиотрофия Кеннеди, подробнее рассмотрен ниже.
При большинстве форм аутосомно-доминантных атаксий с экспансией три- либо пентануклеотидных повторов наблюдается ряд весьма типичных клинико-генетических характеристик, свойственных вообще заболеваниям с «динамическими» мутациями (Иллариошкин С.Н. и др., Bricc А., Klockgether Т., Dichgans J., Rosenberg R., Stevanin G. et al.). К ним относятся:
а) обратная корреляция между степенью экспансии (т.е. числом повторов в мутантном аллеле) и возрастом манифестации симптомов болезни;
б) прямая взаимосвязь между степенью экспансии повторов и тяжестью клинических проявлений (у больных с большим числом повторов в соответствующем гене наблюдается тенденция к развитию наиболее «злокачественных» форм заболевания с быстрым прогрессированием и присоединением ряда дополнительных симптомов);
в) феномен антиципации (появление все более тяжелых и ранних случаев болезни в каждом последующем поколении) — обусловлен нестабильностью повтора и нарастанием его длины при передаче мутантного гена от родителя потомкам;
г) эффект «отцовской передачи» (манифестация более ранних и более тяжелых случаев болезни у потомков больного отца) — обусловлен преимущественным удлинением мутантного повтора в мужском гаметогснсзс, тогда как при передаче гена от матери область повтора обычно остается стабильной; этот эффект выражен в родословной особенно резко, если передача гена по мужской линии происходит в нескольких поколениях подряд;
д) происхождение мутаций de novo от имеющихся в популяции редких аллелей с «промежуточным» числом повторов. Такие аллели сами по себе не приводят к болезни, но являются генетически нестабильными и способны переходить в «полную мутацию» при передаче гена потомкам. Прямое доказательство существования «промежуточных» аллелей было представлено, в частности, для СЦА7 (Stevanin G. et al., Giunti P. et al.).
Механизм генетической нестабильности мутантного аллеля при аутосомно-доминантных атаксиях имеет особую природу. Показано, что у больных СЦА1 и СЦА2 мутантный ген содержит непрерывный (CAG)n-повтор, тогда как нормальные аллели генов прерываются отдельными вставками других триплетов (Chung М. et al., Sanpei К. et al.). Предполагается, что эти вставки служат стабилизирующим фактором, случайная утрата которого и образование непрерывной тандемной конфигурации ведут к появлению генетически нестабильного мутантного аллеля (Zoghbi Н.).
Учитывая большое клинико-генетическое сходство прогрессирующих аутосомно-доминантных спиноцеребеллярных атаксий между собой, еще несколько лет назад многие авторы считали динамическую экспансию тандемных повторов универсальным типом мутаций для этих болезней (или, по крайней мере, для всех форм, при которых имеется феномен антиципации — например, для СЦА5, СЦА11 и др.) (Zoghbi Н., Klockgether Т., Dichgans J., Worth P. et al.).
Однако оказалось, что это не так. В начале нового столетия были идентифицированы 4 формы прогрессирующих аутосомно-доминантных спиноцеребеллярных атаксий (СЦА4, СЦА5, СЦА14 и СЦА27), которые никак не связаны с экспансией повторов и обусловлены вполне «традиционными» точковыми мутациями в генах PLEKHG4, SPTBN2, PRKCG и FGF14 (Chen D.H. et al., van Swicten J.С. et al., Ishikawa K. et al., Ikeda Y. et al.).
Более того, при СЦА4, СЦА5 и СЦА14 ранее был описан феномен антиципации, позволявший обсуждать возможность «динамических» мутаций. Таким образом, клонирование генов PLEKHG4, SPTBN2, PRKCG и открытие в них мутаций по типу нуклеотидных замен ясно демонстрирует, что механизмы «утяжеления» и «омоложения» болезни в ряду последовательных поколений отнюдь не исчерпываются экспансией простых тандемных повторов, а могут иметь под собой и какие-то иные, не вполне пока известные, причины.
Клеточная функция белковых продуктов большинства генов прогрессирующих аутосомно-доминантных спиноцеребеллярных атаксий (атаксины-1, -2, -3 и т.д., атрофии) до настоящего времени не установлена. Напротив, для форм атаксий СЦА6, СЦА12, СЦА14, СЦА17 и СЦА27 функция белков известна; продуктами этих генов являются соответственно а1А-субъсдиница потенциалзависимого кальциевого канала (Zhuchenko О. et al.), регуляторная субъединица белковой фосфатазы РР2А (Holmes S. et al.), протеинкиназа С-у (белок PRKCG) (Chen D.H. et al.), фактор транскрипции TBP (Nakamura К. et al.) и фактор роста фибробластов-14 (белок FGF14) (van Swieten J.С. et al.).
Степень экспансии тринуклеотидных повторов наиболее выражена при СЦА7; эта же форма характеризуется чрезвычайно высокой нестабильностью мутантного аллеля при передаче гена в следующее поколение. Описаны (при отцовской передаче мутации) крайне тяжелые врожденные случаи СЦА7 с гибелью больных в течение первых нескольких месяцев жизни, обусловленные экстремальной экспансией CAG-повторов до 250—300 копий (Benton С. et al., Johansson J. et al.).
С другой стороны, патологические аллели гена а1А-потенциалзависимого кальциевого канала (CACNL1A4), ответственные за развитие СЦА6, генетически весьма стабильны и характеризуются наименьшим среди всех полиглутаминовых заболеваний порогом числа тандемных повторов, приводящим к болезни. Генетические характеристики мутантных аллелей у больных СЦА1, СЦА2, СЦАЗ, СЦА12, СЦА17 и ДРПЛА являются в значительной степени сходными.
Интересно отметить, что в описанных редчайших случаях гомозиготности по мутациям СЦА1 и СЦА6 клиническая картина болезни не отличалась от обычных случаев, обусловленных экспансией CAG в одном аллеле гена (Goldfarb L. et al., Matsuyama Z. et al.); в то же время, у гомозигот по мутации СЦА3 двойная доза мутантного гена характеризовалась «аддитивным» эффектом и приводила к более раннему и более неблагоприятному варианту заболевания (Lang A. et al., Kawakami Н. et al.).
- Читать "Эпидемиология аутосомно-доминантных атаксий - распространенность, частота"
Оглавление темы "Аутосомно-доминантные атаксии":- Аутосомно-доминантные атаксии - история изучения
- Классификация аутосомно-доминантных атаксий
- Генетика аутосомно-доминантных атаксий
- Эпидемиология аутосомно-доминантных атаксий - распространенность, частота
- Патогенез полиглутаминовых форм аутосомно-доминантных атаксий
- Патогенез спиноцеребеллярных атаксий - СЦА8, СЦА10 и СЦА12
- Трансгенные модели аутосомно-доминантных атаксий
- Клиника аутосомно-доминантных атаксий и их течение
- Пример аутосомно-доминантной атаксии
- Патоморфология аутосомно-доминантной атаксии - гистология