
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Дифференциальная диагностика идиопатической поздней мозжечковой атаксии
При идиопатической поздней мозжечковой атаксии, как и при других формах атаксий дегенеративной природы, дифференциальный диагноз проводится в первую очередь с большим числом заболеваний, проявляющихся преимущественно нарушением координации движений. Этот дифференциально-диагностический алгоритм подробно рассмотрен в статьях, посвященной аутосомно-доминантным атаксиям.
Принимая во внимание, что при оливопонтоцеребеллярной атрофии (ОПЦА) нередко имеет место сочетание координаторных расстройств с паркинсонизмом и симптомами вовлечения других областей ЦНС, это заболевание необходимо дифференцировать с болезнью Паркинсона и рядом спорадических мультисистемных дегенерации из группы синдромов паркинсонизм-«плюс» — прогрессирующим надъядерным параличом, кортикобазальной дегенерацией, болезнью диффузных телец Леви (деменцией с тельцами Леви) и др.
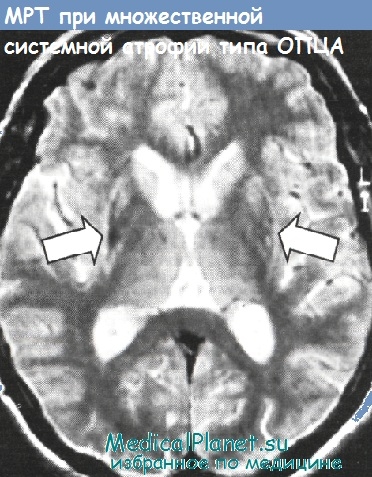
Обычно трудности дифференциальной диагностики ОПЦА-варианта множественной системной атрофии с болезнью Паркинсона возникают лишь в самой ранней стадии болезни и легко разрешаются даже при относительно кратковременном наблюдении за больным на протяжении 6—12 мес: для ОПЦА, дебютирующей синдромом паркинсонизма, типично быстрое присоединение атаксии и других симптомов, отсутствие типичного паркинсоновского тремора покоя, отсутствующий либо минимальный и быстро истощающийся эффект препаратов леводопы, наличие атрофии мозжечка и ствола мозга по данным КТ и МРТ.
В пользу прогрессирующего надъядерного паралича свидетельствует парез вертикального взора (особенно при взгляде вниз), характерная разгибательная поза головы и туловища, быстро нарастающий псевдобульбарный синдром, палилалия, частые падения назад (появляющиеся уже в дебюте болезни), а также заметная атрофия структур среднего мозга на MP-томограммах.
Заподозрить кортикобазальную дегенерацию позволяет наличие у больного апраксии, афазии, феномена «чужой руки», миоклоний, сенсорных расстройств коркового типа, лобных знаков, асимметричной атрофии лобно-теменных областей мозга по данным МРТ. Для болезни диффузных телец Леви весьма характерно раннее развитие деменции, аффективных и психотических расстройств, которые нередко могут быть дебютом болезни, «обрастая» позднее другими разнообразными симптомами. Следует подчеркнуть, что для всех перечисленных нейродегенеративных заболеваний мозжечковая атаксия не характерна (расстройства походки у таких пациентов носят, как правило, сложный лобно-подкорковый характер), поэтому манифестация на фоне мультисистемного процесса «истинных» координаторных нарушений с развитием атрофии мозжечка обычно позволяет врачу четко ориентироваться на диагноз ОПЦА.
Непростой бывает дифференциальная диагностика оливопонтоцеребеллярной атрофии (ОПЦА) с прионными болезнями — болезнью Крейтцфельдта—Якоба и синдромом Герстманна—Штреуслера—Шейнкера, поскольку упомянутые страдания в ряде случаев могут проявляться комбинацией симптомов мозжечковой атаксии и паркинсонизма. В пользу прионной болезни свидетельствует «злокачественное» подострое течение процесса с быстрым (на протяжении нескольких месяцев) присоединением деменции, корковой слепоты, миоклоний, эпилептических припадков, амиотрофий, выявлением на ЭЭГ характерных периодических комплексов типа острая волна—медленная волна, следующих с частотой 1-2 в секунду.
Синдром кортикальной мозжечковой атрофии возможен при многолетнем злоупотреблении алкоголем. Клиническая картина такого синдрома весьма близка к проявлениям идиопатической паренхиматозной кортикальной мозжечковой атрофии и включает выраженную шаткость походки, резкое нарушение координации и интенционный тремор в ногах (функция рук страдает в значительно меньшей степени), негрубую дизартрию, признаки полинейропатии (угнетение сухожильных рефлексов, чувствительные расстройства, боли и др.), тремор пгшьцсв рук.
Характерно подострое развитие симптоматики, нередко приуроченное к очередному запою, после чего в дальнейшем заболевание прогрессирует очень медленно и не влияет на продолжительность жизни. Обычно после прекращения приема алкоголя нарастание симптомов приостанавливается, однако существенного уменьшения проявлений болезни не отмечается. При КТ- и МРТ-исследовании у таких пациентов, как правило, обнаруживается не только атрофия червя и полушарий мозжечка, но и заметное расширение субарахноидальных пространств больших полушарий мозга.
Дегенеративные изменения мозжечка, напоминающие паренхиматозную кортикальную мозжечковую атрофию, могут быть осложнением ряда других патологических процессов. Они описаны при злокачественных новообразованиях (рак легкого и бронхов, рак яичников, матки, лимфома, болезнь Ходжкина и др.), заболеваниях щитовидной железы (гипотиреоз), длительном приеме антиконвульсантов, интоксикации тяжелыми металлами, энтеритах, тепловом ударе и др. (Greenfield J.G., Mancall E.L.). Общим для этой группы синдромов отличием от «истинной» кортикальной мозжечковой атрофии является то, что патологический процесс в коре мозжечка носит распространенный, диффузный характер — поражаются все слои palaeo- и neocerebellum. Основная неврологическая особенность указанных синдромов — подостро развивающаяся статическая и локомоторная атаксия.
При обследовании больных с подозрением на идиопатическую атаксию необходимо исключать также сравнительно недавно описанное заболевание аутоиммунной природы — так называемую глютеновую атаксию. Глютеновая атаксия является следствием непереносимости некоторых компонентов пищевого глютсна (глиадина, проламинов), содержащихся в злаковых культурах — пшенице, ячмене, ржи. В общей выборке спорадических атаксий неясного генеза глютеновая атаксия выявляется в среднем по разным популяциям у 11 — 15% больных, что требует постоянной настороженности в отношении данного заболевания (Hadjivassiliou М. et al., Burk К. et al.).
Симптоматика глютеновой атаксии может проявляться в весьма широком возрастном интервале, но в среднем в 40-50 лет. Характерно развитие сравнительно нсгрубой статико-локомоторной атаксии и лишь незначительной дискоординации в конечностях, реже — дисфагии, расстройств тазовых функций, нарушений глубокой чувствительности, угнетения ахилловых рефлексов, фасцикуляций и амиотрофий. Примерно у 1/4 пациентов отмечается скрытая или симптоматическая глютеновая энтеропатия (могут наблюдаться диарея, синдром мальабсорбции, потеря массы и т.п., изменения слизистой оболочки тонкого кишечника при дуоденальной биопсии). На КТ/МРТ при глютсновой атаксии отмечается атрофия полушарий и червя мозжечка при сохранности структур ствола мозга. При ЭНМГ-исслсдовании могут выявляться признаки аксональной невропатии (сенсорной, моторной или смешанной).
Больные глютсновой атаксией являются носителями определенных гаплотипов главного комплекса гистосовместимости — HLA DQ2 (90% пациентов) или HLA DR4 DQ8 (10%), поэтому результаты иммуногенетического обследования имеют определенную диагностическую ценность (Biirk К. et al.). Основное же значение в диагностике глютсновой атаксии имеет обнаружение в крови больных антител к глиадину и эндомизию. Следует подчеркнуть, что своевременное выявление глютсновой атаксии крайне важно для назначения специальной безглютеновой диеты: показано, что раннее назначение такой диеты может привести к полному исчезновению симптоматики либо как минимум к предотвращению дальнейшего прогрессировать болезни.
Во всех случаях предполагаемой идиопатической поздней мозжечковой атаксии дифференциальный диагноз следует проводить с разнообразными генокопиями, т.е. с наследственными формами атаксий у тех пациентов, у кого в силу различных причин не удается выявить семейный анамнез болезни. Например, в случае возникновения новой мутации и том или ином гене аутосомно-доминантных атаксий у соответствующего больного будет отсутствовать семейный анамнез, однако в дальнейшем заболевание может быть передано потомкам в следующие поколения.
Видимое отсутствие семейного анамнеза при наличии генной мутации может быть связано также с ложным отцовством или ранней смертью одного из родителей, являвшегося носителем мутации и не дожившего до обычного возраста манифестации болезни. В некоторых случаях пациент может просто не располагать какими-либо сведениями о родителях (усыновление, воспитание в детском доме и т.п.). Еще одним показательным примером ошибочной диагностики идиопатической поздней мозжечковой атаксии могут служить поздние случаи аутосомно-рецессивных наследственных атаксий, поскольку при таком типе наследования в семье нередко выявляется лишь один больной. В частности, атипичные поздние варианты атаксии Фридрейха, связанные с «мягкими» мутациями в гене FRDA, нередко представляют значительные диагностические трудности и могут быть ошибочно квалифицированы как случаи идиопатической мозжечковой атаксии.
Некоторые особенности клинической картины позволяют даже при отсутствии семейного анамнеза заподозрить именно наследственную форму атаксии и предпринять необходимые шаги с целью более точной молекулярной диагностики. Например, атрофия зрительных нервов, офтальмоплегия и дегенерация сетчатки встречаются почти исключительно в семейных (аутосомно-доминантных) случаях атаксий и совершенно не характерны для идиопатической спорадической поздней атаксии (Harding А.Е.). Предположение о возможности атаксии Фридрейха должно быть сделано при наличии у больного тотальной сухожильной арефлексии, выраженных нарушений глубокой чувствительности или метаболической кардиомиопатии.
Наличие у пожилого больного двусторонних очагов в белом веществе средних ножек мозжечка весьма характерно для синдрома FXTAS. Однако в большинстве случаев какие-либо специфические симптомы, позволяющие дифференцировать наследственные и «истинные» спорадические формы атаксий, отсутствуют.
Таким образом, достоверное вычленение идиопатической мозжечковой атаксии из общей группы спорадических мозжечковых дегенеративных синдромов, часть из которых могут иметь наследственную природу, возможно только на основании прямой ДНК-диагностики и тотального мутационного скрининга, позволяющего исключить мутации в соответствующих генах-кандидатах.
- Читать "Пример идиопатической поздней мозжечковой атаксии"
Оглавление темы "Идиопатические атаксии":- Атаксия при метаболических лейкодистрофиях - клиника, диагностика
- Атаксия при врожденных болезнях гликозилирования - клиника, диагностика
- Идиопатическая поздняя мозжечковая атаксия - определение, история изучения
- Классификация идиопатической поздней мозжечковой атаксии
- Оливопонтоцеребеллярная атрофия (ОПЦА) - клиника, диагностика
- Морфология оливопонтоцеребеллярной атрофии (ОПЦА)
- Паренхиматозная кортикальная мозжечковая атрофия - клиника, диагностика
- Диагностика идиопатической поздней мозжечковой атаксии
- Дифференциальная диагностика идиопатической поздней мозжечковой атаксии
- Пример идиопатической поздней мозжечковой атаксии