
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Диагностика синдрома FXTAS. ДНК-анализ
Из всех дополнительных лабораторно-инструментальных методов исследования при синдроме FXTAS наибольшее диагностическое значение (не считая ДНК-тестирования) имеет МРТ головного мозга. Практически у всех больных выявляется довольно характерный и редкий на практике паттерн: двусторонние очаги повышенной интенсивности сигнала (в режиме Т2) в области средних ножек мозжечка, реже — нижнего ствола, в комбинации с очагами в прилегающих к средним ножкам глубоких отделах белого вещества полушарий мозжечка, но всегда с сохранностью зубчатых ядер (Brunberg J.A. et al., Jacquemont S. et al.).
Менее специфичными являются признаки корковой атрофии мозжечка и большого мозга, особенно в лобных и теменных отделах, а также очаговые изменения в перивентрикулярных и глубоких отделах белого вещества больших полушарий (в некоторых случаях это было основанием для первоначального некорректного диагноза «болезнь Бинсвангера»), истончение мозолистого тела. При электронейромиографическом исследовании нередко выявляются признаки аксональной дистальной полиневропатии, реже — вовлечения в процесс мотонейронов спинного мозга.
Для облегчения клинической диагностики синдрома FXTAS предложены критерии «достоверного», «вероятного» и «возможного» диагноза данного заболевания, основанные на комбинации «больших» и «малых» клинико-радиологических признаков (Jacquemont S. et al.). Эти признаки перечислены в таблице ниже. В соответствии с ними критерии диагностики синдрома FXTAS следующие:
1. «Достоверный» диагноз — наличие у больного одного «большого» клинического и одного «большого» радиологического признака.
2. «Вероятный» диагноз — наличие двух «больших» клинических признаков либо наличие одного «большого» радиологического + одного «малого» клинического признака.
3. «Возможный» диагноз — наличие одного «большого» клинического признака + одного «малого» радиологического признака.
Эти критерии носят универсальный характер и применимы для диагностики синдрома FXTAS как у мужчин, так и у женщин (Hagerman R.J. et al.).
«Большие» и «малые» признаки клинико-радиологической диагностики синдрома FXTAS (по S.Jacquemont и соавт.)
| Признаки и их категоризация | Описание признака |
|
Радиологические
«большой» «малый» «малый» |
МРТ-очаги в белом веществе средних ножек мозжечка и/или ствола мозга МРТ-очаги в белом веществе больших полушарий мозга Умеренная либо выраженная генерализованная атрофия мозга |
|
Клинические
«большой» «большой» «малый» «малый» «малый» |
Интенционный тремор Атаксия ходьбы Паркинсонизм Умеренный либо выраженный дефицит кратковременной памяти Нарушения экзекутивных функций |
Дифференциальный диагноз при синдроме FXTAS следует проводить с множественной системной атрофией и в особенности ее оливопонтоцеребеллярным фенотипом, паренхиматозной кортикальной мозжечковой атрофией, аутосомно-доминантными атаксиями, болезнью Паркинсона и разнообразными нейродегенеративными заболеваниями позднего возраста из группы «паркинсонизм-плюс» (прогрессирующим надъядерным параличом, деменцией с тельцами Леви, лобно-височной деменцией с паркинсонизмом и др.), болезнью Бинсвангера и другими формами хронической цереброваскулярной недостаточности, системными васкулитами позднего возраста, болезнью Альцгеймера, болезнью Гентингтона, атипичными вариантами эссенциального тремора, паранеопластическими нейродегенеративными процессами.
Во всех случаях ключом к правильной диагностике является тщательно собранный семейный анамнез (наличие случаев умственной отсталости у племянников по женской линии или внуков, наличие нейродегенеративных заболеваний у братьев или других пожилых родственников по женской линии), особенности неврологического синдрома и данные МРТ (очаги в средних мозжечковых ножках в сочетании с другими очаговыми или атрофическими изменениями головного мозга).
Разумеется, ведущая роль в диагностике синдрома FXTAS принадлежит прямому ДНК-тестированию. Традиционный ДНК-анализ тандсмной последовательности CGG в гене FMR1 предполагает проведение Саузерн-блот-гибридизации с использованием рестриктаз EcoRI и NruI и зонда StB 12.3.
Однако сравнительно небольшой размер экспансии CGG-повторов в «промежуточном» аллеле гена у больных FXTAS позволяет значительно упростить ДНК-анализ благодаря использованию полимеразной цепной реакции (ПЦР). Более того, даже при обнаружении мутантного аллеля с помощью блот-гибридизации точный количественный анализ числа CGG-повторов возможен только на основе ПЦР.
Как и для большинства других «болезней экспансии», ПЦР-диагностика синдрома FXTAS дает достаточную амплификацию и четкое разделение нормальных и мутантных аллелей, что позволяет быстро (в течение одного дня) выявлять носительство мутации в любом конкретном случае на клинической, доклинической и пренатальной стадиях.
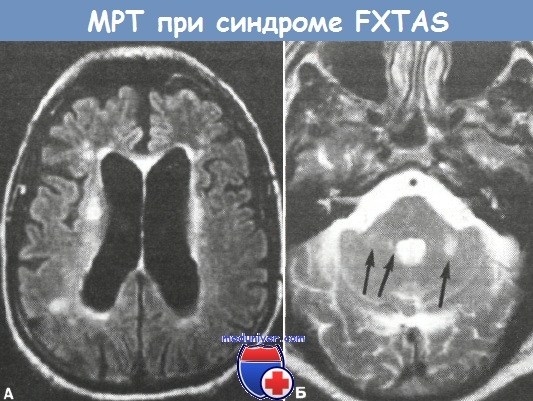
а — очаговые изменения в перивентрикулярных отделах белого вещества больших полушарий;
б — очаги повышенной интенсивности сигнала в области средних ножек мозжечка (короткие стрелки)
- Читать "Лечение и профилактика синдрома FXTAS. Пренатальная диагностика"
Оглавление темы "Наследственные Х-сцепленные рецессивные атаксии":- Спиноцеребеллярная атаксия с аномальными саккадами — синдром SCASI
- Х-сцепленные рецессивные атаксии - история изучения, классификация
- Синдром FXTAS - причины, генетика
- Клиника и патогенез синдрома FXTAS
- Диагностика синдрома FXTAS. ДНК-анализ
- Лечение и профилактика синдрома FXTAS. Пренатальная диагностика
- Фридрейхоподобная Х-сцепленная атаксия - причины, клиника, диагностика
- Фатальная Х-сцепленная атаксия с глухотой и слепотой — синдром Артса
- X-сцепленные формы спастической атаксии - причины, клиника, диагностика
- X-сцепленная атаксия с поздней деменцией - причины, клиника, диагностика