
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Дентаторубро-паллидолюисова атрофия (ДРПЛА) - клиника, диагностика
Дентаторубро-паллидолюисова атрофия (ДРПЛА, MIM 125370) - форма доминантных атаксий (ее иногда относят также к группе наследственных деменций или хореических синдромов) — чрезвычайно редкое популяционно-специфичное заболевание, которое встречается почти исключительно в Японии с частотой 1 на 1 000 000.
Единичные случаи дентаторубро-паллидолюисова атрофии описаны в Европе и Северной Америке (Warner Т. et al.).
Симптоматика дентаторубро-паллидолюисовой атрофии чрезвычайно полиморфна. Заболевание может начинаться в возрасте от 1-го до 6-го десятилетия жизни и проявляется в виде хорсоатетоза, атаксии, деменций, миоклоний, эпилептических припадков (Naito Н., Oyanagi S., Iizuka R. et al.).
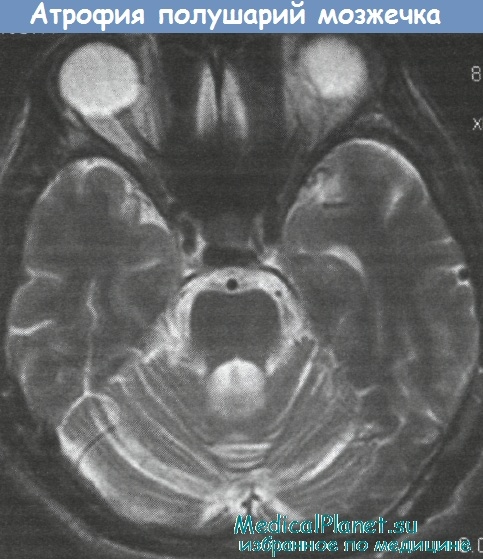
Важным диагностическим признаком дентаторубро-паллидолюисовой атрофии является выявление на MP-томограммах головного мозга, помимо неспецифических атрофических изменений мозжечка, ствола мозга и больших полушарий, очагов демиелинизации в белом веществе перивентрикулярной области и ссмиовального центра больших полушарий. В семьях, отягощенных дентаторубро-паллидолюисовой атрофии, нередко наблюдаются антиципация и феномен «отцовской передачи».
Продолжительность болезни обычно не превышает 15 лет. Морфологически ДРПЛА характеризуется дегенеративными изменениями в зубчатом ядре, наружном сегменте бледного шара и их проекционных зонах в красном и люисовом ядрах, атрофией коры больших полушарий (Iizuka R. et al.), а также наличием внутриядерных белковых включений, содержащих мутантный продукт гена с удлиненной полиглутаминовой цепью (Becher М. et al.).
Дентаторубро-паллидолюисова атрофия является типичным представителем полиглутаминовых болезней. Ген ДРПЛА, локализованный на хромосоме 12р12—ter и кодирующий белок с неустановленной функцией (атрофии), содержит участок тандемных CAG-повторов, число которых в норме составляет < 36, а на мутантных хромосомах — от 49 до 88 (Koide R. et al.).
Анализ клинико-генетических корреляций показал, что различная степень экспансии CAG-повторов мутантного гена приводит к манифестации двух различных по клиническому синдрому и тяжести фенотипов болезни.
При небольшой степени экспансии CAG-повторов наблюдаются более поздний дебют и развитие хорсоатетоза, атаксии, психических нарушений (данный фенотип иногда обозначается в литературе как «псевдохорея»), в то время как у больных с максимальной длиной повтора болезнь проявляется в более раннем возрасте тяжелым синдромом прогрессирующей миоклонус-эпилепсии и деменцией (Ikeuchi Т. et al.).
- Читать "Дифференциация спиноцеребеллярных атаксий"
Оглавление темы "Прогрессирующие спиноцеребеллярные атаксии (СЦА)":- Спиноцеребеллярная атаксия 20-го типа (СЦА20) - клиника, диагностика
- Спиноцеребеллярная атаксия 21-го типа (СЦА21) - клиника, диагностика
- Спиноцеребеллярная атаксия 22-го типа (СЦА22) - клиника, диагностика
- Спиноцеребеллярная атаксия 23-го типа (СЦА23) - клиника, диагностика
- Спиноцеребеллярная атаксия 25-го типа (СЦА25) - клиника, диагностика
- Спиноцеребеллярная атаксия 26-го типа (СЦА26) - клиника, диагностика
- Спиноцеребеллярная атаксия 27-го типа (СЦА27) - клиника, диагностика
- Спиноцеребеллярная атаксия 28-го типа (СЦА28) - клиника, диагностика
- Дентаторубро-паллидолюисова атрофия (ДРПЛА) - клиника, диагностика
- Дифференциация спиноцеребеллярных атаксий