
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Дефекты молекулярного транспорта в нейронах при НСП - KIF5A, спастин, атластин, спартин
Дефекты молекулярного транспорта в нейронах - KIF5A, спастин, атластин, спартин и др. в последнее время привлекают особое внимание исследователей. По мере идентификации новых генов появляется все больше данных о связи нейродегенерации при НСП с нарушениями транспортных функций соответствующих нейрональных белков.
Такая связь наиболее отчетлива для формы SPG10, связанной с мутациями гена KIF5A. Его продукт принадлежит к обширному семейству кинезинов — молекулярных «двигателей», осуществляющих транспорт жизненно важных субстратов по микротрубочкам цитоскелета при участии АТФ. У человека известно свыше 40 различных «двигательных» кинезинов. Белок KIF5A экспрсссируется почти исключительно в мотонейронах и является компонентом сложного кинезинового гетеротетрамерного комплекса, в состав которого входят:
а) две субъединицы тяжелой цепи кинезина (у млекопитающих она представлена белками KIF5A, KIF5B или KIF5C);
б) две субъединицы легкой цепи кинезина (белки KLC1, KLC2 или KLC3). Этот комплекс играет роль «мотора», перемещающего органические субстраты к периферии клетки (в нейронах это быстрый антероград-ный аксональный транспорт).
Точная субстратная специфичность кинезинов не до конца известна, однако они, наиболее вероятно, причастны к транспорту митохондрий, лизосом, эндосом, белковых комплексов и мРНК в клетке, а также к мембранному переносу в системе «эндоплазматический ретикулум — аппарат Гольджи» (Reid Е.). Поскольку в культуре клеток мышей с отсутствием K.IF5B обнаруживается аномальное распределение митохондрий и лизосом, можно предположить, что и близкий белок KIF5A также ответственен за транспорт именно этих органелл в нейронах.
«Кинезиновая» модель наследственной спастической параплегии (НСП) находит и экспериментальное подтверждение: мутации гомологичных генов тяжелой цепи кинезинов у ряда низших организмов (мышей и др.) вызывают двигательные нарушения, сходные с НСП (Rcid Е. et al.).
Различные кинезины связаны также и с другими наследственными болезнями нервной системы: например, генетические дефекты кинезина KIF1Bb вызывают наследственную моторно-сенсорную нейропатию типа 2А, в основе которой также лежит аксонопатия, причем аналогичное заболевание есть и у мышей (Zhao С. et al.). Двигательные нарушения у мышей возникают и в результате мутаций легкой цепи кинезина (Rahman A. et al.). Таким образом, на сегодняшний день можно говорить о «кинезинопатиях» как о новом растущем классе нейродегенеративных заболеваний, связанных с нарушением транспортных функций аксонального цитоскелета как в периферических, так и в центральных нейронах.
С транспортными функциями связывают в клетке и роль белка спастина, ответственного за SPG4 — самую частую доминантную форму НСП. При этом основное значение придается способности N-концевого участка молекулы спастина связываться с микротрубочками цитоскелета — центральным элементом транспортной системы клетки. Диссоциация спастина и микротрубочек является АТФ-зависимым процессом, а самому спастину приписывают свойства микротубулоразъединяющего фермента (подобно некоторым другим AAA-белкам - катанину и др.) (Crosby A., Proukakis С., Reid Е.). Таким образом, спастин определенным образом обеспечивает нормальную кинетику микротубулярного аппарата клетки при осуществлении им транспорта необходимых субстратов.
Предполагается, что большинство нарушений структуры спастина при SPG4 реализуется по механизму «доминантно-негативных» мутаций: при условии сохранной N-концевой части мутантные молекулы сохраняют способность к связыванию с микротрубочками и тем самым блокируют сайты связывания для нормальных молекул спастина или иных спастин-ассоциированных белков (Reid Е.). Результатом этих мутаций является нарушение организации, стабильности и числа микротрубочек (что подтверждается в эксперименте на трансгенных дрозофилах с инактивированным спасти-ном) с последующей патологией аксонального транспорта и ретроградной гибелью протяженных пирамидных путей (Sherwood N. et al., Trotta N. et al.).
Механизмы действия атластина, ответственного за развитие SPG3, и спартина, с которым связана форма SPG20 (классический синдром Тройер), до настоящего времени остаются лишь предположительными.
Атластину отводят важную роль в обеспечении синаптического транспорта. Данный белок, широко экспрессирующийся в головном и спинном мозге, имеет структурную гомологию с ГТФазами, принадлежащими к семейству динаминов, и на основании экспериментальных данных может быть отнесен к этой группе. Динамины же играют важнейшую роль в нескольких этапах синаптического транспорта, обеспечивающего нейротрансмиссию, нейротрофическое действие и рециркуляцию синаптических везикул, а также участвуют в дисперсии митохондрий и связывании с микротрубочками цитоскелета (McNiven М. et al., Zhao X. et al., Zhu P. et al., 2003). Можно предположить, таким образом, транспортирующую и «митохондриальную» роль атластина в пирамидных нейронах.
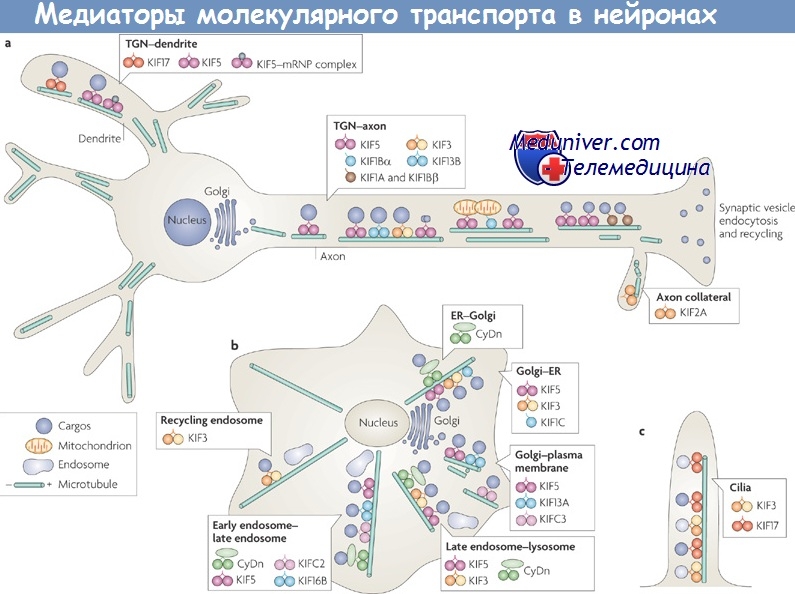
При трех аллельных НСП-подобных аутосомно-рецессивных заболеваниях, связанных с мутациями гена ALS2 на хромосоме 2q33 (раннем восходящем спастическом параличе и др.), генетический дефект затрагивает ГТФазоподобный белок алсин, гомологичной атластину и предположительно выполняющий в клетке сходные функции.
Что касается спартина, то на его потенциальную роль в транспортных процессах косвенно указывает гомология с некоторыми белками, участвующими в эндосомальной морфологии и транспорте поздних эндосомальных компонентов (SNX15, VPS4, SK.D1, RPK1) (Patel Н. et al.). Еще более важной с этой точки зрения является недавно установленная гомология спартина с рассмотренным выше белком спастином — и именно с его N-концевым участком, обеспечивающим связывание с микротрубочками (Ciccarelli F. et al.).
Транспортными белками предположительно являются также продукты последних идентифицированных генов НСП: NIPAI (форма SPG6) и маспардин (SPG21), однако более детальный анализ функции этих новых белков пока не проведен.
Таким образом, у KIF5A, спастина, атластина, алеина и спартина есть ряд объединяющих свойств, позволяющих сделать определенные важные обобщения. К этим свойствам относятся:
а) способность связываться с микротрубочками (для KIF5A и спастина — доказанная; для атластина, алсина и спартина — предположительная, в силу их гомологии с другими белками, имеющими эту функцию);
б) участие во внутриклеточном транспорте (для KIF5A эта роль несомненна; для атластина, алсина и спартина она предполагается, опять же, на основании гомологии с соответствующими транспортными белками).
Дальнейшие исследования должны выяснить функциональную общность рассмотренных белков и, возможно, единый или близкий механизм дегенерации пирамидных путей при указанных формах спастической параплегии - SPG3, SPG4, SPG6, SPGIO, SPG20, SPG21, а также раннем восходящем спастическом параличе и аллельных ему синдромах (Crosby A., Proukakis С., Reid Е.).
Если рассмотреть вопрос о механизмах развития НСП еще шире—с учетом значимости митохондрий в энергообеспечении АТФ-зависимых процессов субстратной циркуляции в нейронах (формы SPG7 и SPG13) и токсической роли нарушенного аксонального транспорта в созревании миелина (форма SPG2) — можно заключить, что подавляющее большинство форм НСП с известными молекулярными дефектами представляют собой болезни аксонального транспорта (Crosby A., Proukakis С.).
Поскольку аксоны пирамидного тракта являются в организме наиболее протяженными (их длина у человека может достигать 1 м), именно в этих клетках аксональный транспорт является наиболее сложно организованным и ранимым процессом, поддержание которого представляет особенно серьезную проблему, критическую для судьбы нейрона. Вот почему мутации повсеместно экспрессирующихся генов (таких, например, как спастин или спартин) вызывают такую избирательную дегенерацию именно кортикоспинальных аксонов. При этом, естественно, наиболее чувствительными к патологии аксонального транспорта являются самые отдаленные от тела нейрона участки аксонов, от которых дегенеративный процесс постепенно распространяется более проксимально — от периферии к центру нейрона (гибель нейрона по типу «dying back»).
Интересно отметить, что ряд наиболее часто встречающихся при НСП дополнительных симптомов (нарушение глубокой чувствительности, мозжечковая недостаточность, проявления периферической полиневропатии) также связаны с поражением чрезвычайно длинных и, следовательно, особенно ранимых при дефектах аксонального транспорта нервных волокон — в воставс задних столбов, спиноцеребеллярных трактов или периферических нервов.
Как справедливо отмечает E.Reid, по мерс идентификации генов НСП и изучения механизмов их действия на первый план выходит целый ряд важнейших вопросов:
• Какие еще факторы (помимо рассмотренных выше) могут вносить вклад в избирательную чувствительность кортикоспи-нальных аксонов к изменениям белков, экспрессия которых в организме весьма широка?
• Какую роль играет длина аксонов в этих процессах?
• Каковы молекулярные механизмы возрастной зависимости и внутрисемейных клинических различий конкретных форм НСП?
• Какие факторы «запускают» формы с поздним началом?
Исследования в указанных направлениях активно проводятся в ведущих лабораториях мира; в частности, уже есть предварительные данные о возможных генах-модификаторах при НСП, существенно влияющих на фенотипические проявления данных заболеваний (Meyer Т. et al.). Дальнейший анализ этих факторов, по-видимому, станет в ближайшие годы одним из актуальных направлений нейрогенетики, потенциально позволяющим рассчитывать на разработку подходов к принципиально новым, патогенетическим методам лечения НСП.
- Читать "Спастическая параплегия с эпилепсией и умственной отсталостью - клиника, диагностика"
Оглавление темы "Осложненные наследственные спастические параплегии (НСП)":- Дефекты молекулярного транспорта в нейронах при НСП - KIF5A, спастин, атластин, спартин
- Спастическая параплегия с эпилепсией и умственной отсталостью - клиника, диагностика
- Спастическая параплегия с экстрапирамидными расстройствами - клиника, диагностика
- Спастическая параплегия, нейросенсорная глухота, умственная отсталость и прогрессирующая нефропатия - клиника, диагностика
- Редкие формы доминантных наследственных спастических параплегий (НСП)
- Спастическая параплегия с макроцефалией и лицевым дисморфизмом - синдром Фринса
- Спастическая параплегия с брахидактилией типа Е - синдром Фицсиммонса-Гилберта
- Спастическая параплегия с миоклонической эпилепсией
- Спастическая параплегия с эктродактилией и умственной отсталостью - синдром Дженкара
- Спастическая параплегия с синдромом Эванса - клиника, диагностика