
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Генетический контроль половой дифференцировки - половые хромосомы
Первый этап открытия TDF состоял в определении локализации гена (или генов) на Y-хромосоме. Этому способствовало исследование больных с аберрациями Y-хромосомы. Прежде всего было найдено, что TDF картируется на коротком плече Y-хромосомы (Yp). Об этом свидетельствовали наблюдения, согласно которым пациенты с кариотипом 46X,i(Yq) имели женский фенотип. У таких пациентов имелась Y-хромосома с двумя длинными (Yq) плечами вместо обычных одного короткого (р) и одного длинного (q) плеч. По существу имела место делеция короткого плеча.
Позже удалось исключить конец короткого плеча как район локализации гена, поскольку пациенты с кольцевой Y-хромосомой были мужчинами.
При образовании кольцевой хромосомы концевые части короткого и длинного плеч делетируются, их «липкие» концы смыкаются, образуя кольцо. Эти наблюдения соответствовали тому факту, что в процессе мейоза мужских половых клеток происходит конъюгация Х-хромосомы и Y-хромосомы с обменом генетического материала. Этот участок обязательной конъюгации известен как псевдоаутосомный район и в его состав входит дистальная часть Yp. Вряд ли можно было бы думать о том, что эта область содержит ген(ы), определяющий пол, поскольку любой процесс кроссинговера привел бы к переносу гена TDF на Х-хромосому!
Поэтому линия раздела между конъюгирующим и неконъюгирующим участками Y-хромосомы (псевдоаутосомная фаница) представляет собой дистальную фаницу участка хромосомы, определяющего мужской пол. Исследования, проведенные на других пациентах с терминальными делециями Yp, дали возможность определить проксимальную фаницу этой области, т.е. расположенную наиболее дистально точку разрыва в неконъюгирующем участке, у пациентов, имеющих женский фенотип.
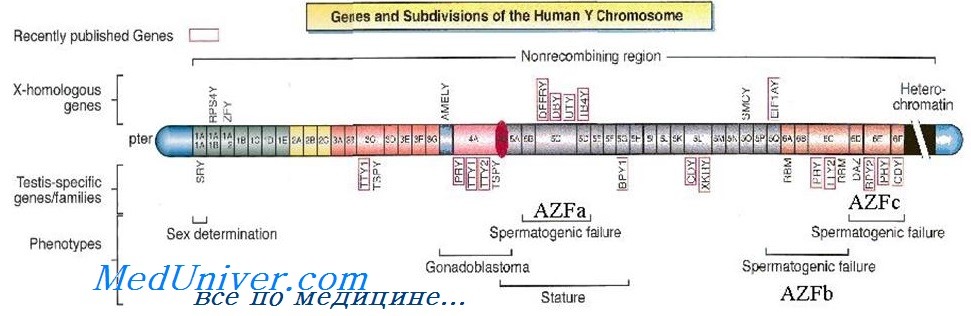
Таким способом удалось сузить фаницы искомой области до полосы Yp11.2, и это означало, что один из генов-кандидатов, ген H-Y, не является TDF, так как ген H-Y, кодирующий H-Y-антиген, картирован на Yq.
Проведение дальнейших исследований потребовало применения молекулярно-генетических методов. Объектом исследования были пациенты особой категории — лица мужского пола, имеющие кариотип 46ХХ, и женщины с половыми хромосомами XY. Полученные результаты показали, что ДНК большинства мужчин с хромосомами XX содержит Y-специфические последовательности. Каковы же причины этого явления? Известно, что мейотическая рекомбинация в псевдоаутосомной области при мужском мейозе представляет собой обычный процесс, не свойственный оставшейся части Y-хромосомы.
Однако в редких случаях точка обмена расположена проксимальнее, что и приводит к переносу гена TDF на Х-хромосому. Сперматозоиды, несущие такую Х-хромосому, дают начало развитию мужской особи с кариотипом 46ХХ, тогда как сперматозоиды, содержащие Y-хромосому без гена TDF, определяют кариотип 46XY при женском фенотипе. Изучение Y-специфических последовательностей у мужчин с кариотипом 46ХХ и редко встречающихся женщин с кариотипом 46XY, у которых Y-хромосома утратила Y-специфические последовательности, дало возможность обнаружить гены-кандидаты для TDF.
Первый возможный ген-кандидат был описан в 1987 г. [Page et al.] и назван ZFY. Этот ген кодирует активатор транскрипции — белок «цинковых пальцев» (отсюда и сокращение ZFY — zinc finger Y), и оказалось, что гомологичная последовательность имеется на Х-хромосоме (ZFX). Однако вслед за этим появился ряд данных, показавших, что ген ZFY не аналогичен гену TDF:
• были обнаружены Y-позитивные мужчины с кариотипом 46ХХ, у которых ген ZFY отсутствовал;
• ген ZFY имеет аутосомную локализацию у млекопитающих отряда сумчатых, пол которых определяется системой хромосом X—Y;
• найдено, что у мышей ген ZFY экспрессируется только клетками зародышевой линии, но не эмбриональными семенниками, в которых половые клетки отсутствуют. Таким образом, ген ZFY не может участвовать в детерминации семенников, однако, возможно, что он играет важную роль в созревании половых клеток.
Поиски ДНК-последовательностей у лиц мужского пола с кариотипом 46ХХ и отсутствием ZFY продолжались, и в 1990 г. удалось изолировать ген SRY (детерминирующая пол область Y — англ. sex-determining region Y) [Sinclair et al., 1990]. Были получены убедительные данные, свидетельствующие о том, что SRY является геном, определяющим развитие семенников:
• ген SRY Y-специфичен и имеет консервативную природу, что обнаружилось при изучении млекопитающих различных видов;
• он делетирован у мышей-самок с XY-гоносомами (половыми хромосомами);
• он экспрессирован в гонадах эмбрионов мышей-самцов на критической стадии дифференцировки семенников;
• у женщин с XY дисгенезией гонад были идентифицированы de novo мутации гена SRY [Berta et al., Jager et al.];
• после введения гена SRY мышиным эмбрионам с набором хромосом XX рождались самцы с ХХ-гоносомами.
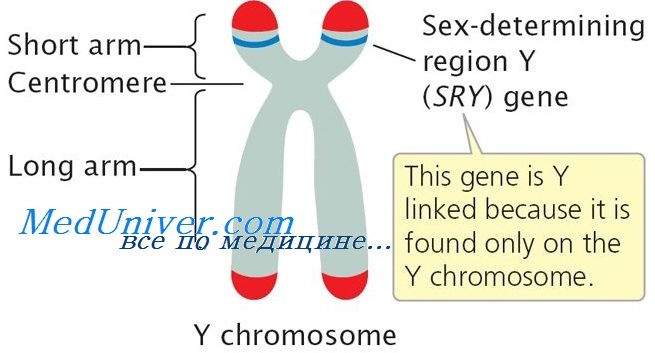
Таким образом, ген SRY действительно является определяющим пол геном, и в настоящее время мутации в этом гене рассматриваются как существенная причина XY реверсии пола. McElreavy и соавт. исследовали 25 пациентов с XY дисгенезией гонад и у 4 обнаружили мутации гена SRY. Еще в одном случае авторы идентифицировали делецию Yp, которая не привела к разрыву гена SRY. Подобное же наблюдение было сделано Capel и соавт., описавшими трех мышей-самок с XY-гоносомами, имеющими различные делеции Yp; однако во всех случаях SRY-локус оставался интактным.
У одной из этих мышей исследовали экспрессию гена SRY, и было обнаружено, что она не определяется на критической стадии развития семенника. Авторы постулировали, что делеция привела к отсутствию экспрессии гена SRY в результате эффекта положения; иными словами, делеция могла сделать ген SRY доступным влиянию центромерного гетерохроматина Y, вследствие чего ген SRY утратил способность экспрессироваться.
Причина развития XY дисгенезии гонад в отсутствие мутаций гена SRY или аномалий Yp неизвестна, однако существуют некоторые данные, позволяющие предположить, что в развитии семенников участвует не только ген SRY. Уникальный случай описан Vilain и соавт. при обследовании одной из семей. Мутация, обнаруженная в гене SRY, обусловила XY реверсию пола у трех женщин этой семьи, однако два других носителя той же мутации были фертильными мужчинами с XY-гоносомами.
Одна из рассматриваемых авторами гипотез состоит в том, что аллель гена SRY мог генерировать два разных фенотипа путем взаимодействия в двумя независимо сегрегирующими аллелями второго гена. В подтверждение гипотезы авторы ссылаются на данные по генетике детерминации пола у мышей, приведенные в работе Eicher и Washburn. В этом обзоре описаны три мыши-самки с XY-хромосомами, за реверсию пола у которых были ответственны аутосомные мутации. У человека не исключено существование гомологичных аутосомных генов, которые могут взаимодействовать с продуктом SRY.
И действительно, исследования, проведенные на Drosophila и нематоде C.elegans [Hodgkin], показывают, что генетический контроль половой дифференцировки у этих видов представляет собой каскадный процесс, в котором участвует серия генов, и действие одного гена последовательно сменяется активным функционированием другого. У C.elegans регуляторный каскад включает 7 взаимодействующих аутосомных генов, контролируемых тремя Х-сцепленными генами [Hunter, Wood]. Возможно, что подобный же процесс происходит и у человека.
Предполагается, что ген SRY кодирует транскрипционный фактор и таким способом контролирует транскрипцию по меньшей мере одного вторичного гена. По-видимому, ген SRY способен не только связываться с ДНК, но и изгибать ее определенным образом. Отсюда можно предположить, что он может обеспечить включение находящихся вслед за ним генов, приближая их к необходимым регуляторным последовательностям.
Наличие генов-мишеней по ходу транскрипции может объяснить существование лиц мужского пола с ХХ-хромосомами, лишенных гена SRY и тем не менее имеющих полный мужской фенотип [Vilain et al.]; сходное объяснение можно дать некоторым случаям истинного гермафродитизма XX, когда у пациентов присутствует и тестикулярная, и овариальная ткань, но отсутствует ген SRY.
Вероятно, у таких пациентов на поздней стадии дифференцировки яичек должна была произойти мутация, обусловливающая «приобретение функции».
McElreavy и соавт. постулируют существование второго гена, названного Z, который действует как негативный регулятор процесса детерминации мужского пола. Авторы предполагают, что ген SRY супрессирует ген Z, давая возможность развития семенников. Рецессивные мутации в гене Z при отсутствии гена SRY будут приводить к формированию особей мужского пола с хромосомами XX, а те мутации в гене Z, которые делают его нечувствительным к действию гена SRY, могут обусловить развитие женского фенотипа при наличии хромосом XY. Гипотетически это можно представить в виде следующей схемы:
Если процесс детерминации пола определяется геном, функционирующим подобно гену Z, или генами с иными функциями, возникает вопрос о природе этих генов и их хромосомной локализации. Данные семейных исследований и анализ спорадических случаев свидетельствуют о возможном участии Х-сцепленного гена. Существуют редкие семьи, в составе которых имеются сибсы, являющиеся Y-негативными истинными гермафродитами, Y-негативными мужчинами с хромосомами XX и нормальными мужчинами и женщинами. Здесь тип наследования соответствует Х-сцепленному — мутация передается через здоровых мужчин или неманифестирующихся женщин, у которых она инактивирована X-хромосомой.
Приводятся также данные о семьях, где дисгенезия гонад, по-видимому, наследуется как Х-сцепленный признак [Sternberg et al., German et al., Mann et al.]; однако в этих случаях возможен аутосомно-доминантный тип наследования с ограничением по мужскому полу.
Еще одно подтверждение наличия Х-сцепленного гена получено в результате изучения случаев реверсии пола у пациентов с XY-гоносомами, имеющих дупликации Х-хромосомы [Bernstein et al., Scherer et al., Stern et al.]. У всех пациентов дупликация была обнаружена в одной и той же области, Хр21/р22. Далее, Ogata и соавт. (1992) приводят описание ребенка с реверсией пола, имеющего кариотип 46X,Yp+, у которого появление Yp обусловлено транслокацией фрагмента Хр (Хр21—22.3) на Yp, что и привело к дупликации этой Хр-области. Существуют и другие случаи XY с дупликациями Хр, где фенотипически пациенты являются мужчинами, т.е. Хр22.1—Хр22.32 [Coles et al.], Хр22.1-Хр22.3 [Narahara et al., 1979] и Xpl 1.2-21.2 [Brondum-Neilson, Langkjaier].
Это показывает, что соответствующий ген, который определяет пол, находится в области Хр21.3 — единственном сегменте, имеющемся у пациентов с женским, но не мужским фенотипом.
На основании данных молекулярно-генетических исследований Ogata и Matsuo приходят к выводу о том, что ген, названный ими TDF-X, может быть расположен между анонимным ДНК-сегментом DXS28 и геном орнитинкарбамилтрансферазы, ОКТ. Bardoni и соавт. (1994) картировали этот локус в регионе Хр21 протяженностью 160 кД, который включает локус, детерминирующий врожденную гипоплазию надпочечников (ВГН). Такой ген может ингибировать дифференцировку по мужскому типу, когда в случае его дупликации оказывается превышенным порог чувствительности гена SRY к ингибирующему влиянию. Поэтому некоторые авторы дали этому локусу другое наименование — DSS (англ. dosage-sensitive sex-reversal — дозочувствительная реверсия пола).
Swain и соавт. предположили, что ген, обозначенный как DAX1, может быть ответственным за фенотип DSS, по крайней мере частично. Функциональная недостаточность этого гена, возникшая вследствие делеций или точковых мутаций, обусловливает у мужчин ВГН и гипогонадотропный гипогонадизм. Исследования на мышах показали, что ген DAX1 экспрессирован в развивающихся гонадах, а также надпочечниках, гипоталамусе и передней доле гипофиза. И действительно, начало его экспрессии в половом валике совпадает по времени с появлением экспрессии гена SRY, а к тому моменту, когда ген SRY уже не экспрессируется, происходит резкое снижение экспрессии гена DAX1 в семеннике при ее сохранении в яичнике. Одно из допустимых объяснений состоит в том, что ген SRY может инициировать в яичках дифференцировку, которая в свою очередь ведет к репрессии гена DAX1.
Высокий уровень экспрессии гена DAX1, как это имеет место при фенотипе DSS, может преодолеть эту репрессию, и не исключено, что у лиц с гоносомами XX этот ген играет существенную роль в развитии яичников.
В процессах, опосредуемых каскадом генов, могут принимать участие другие аутосомные локусы. К такому выводу пришли Simpson и соавт. на основании генетического анализа всех имевшихся к тому времени в литературе данных по XY дисгенезии гонад. В 18 семьях число больных детей было больше одного в семье, а в 3 других семьях изучение родословных указывало на Х-сцепленный или ограниченный по мужскому полу аутосомно-доминантный тип наследования.
Авторы провели сегрегационный анализ в первых 18 семьях, что позволило подсчитать число повторов у мужчин, родившихся после зарегистрированного случая. Полученная в результате анализа величина статистически достоверно не отличалась от ожидаемых 25 % при аутосомно-рецессивном типе наследования по сравнению с 50 % при рецессивном Х-сцепленном наследовании. Имеются также сообщения о случаях XY дисгенезии гонад при кровнородственных браках, что дает дополнительное подтверждение существования аутосомно-рецессивного типа наследования. Сведения о возможной локализации этих аутосомных генов были получены в результате обследования пациентов с реверсией пола, имеющих аномалии аутосом, а также лиц, у которых синдромы, наследуемые по аутосомному типу, возникли вследствие мутаций одиночного гена.
- Вернуться в оглавление раздела "Гинекология"
Оглавление темы "Гинекология":- Бактериальный вагинит у детей - причины, диагностика, лечение
- ВИЧ-инфекция у детей - причины, диагностика, лечение
- Болезни передаваемые половым путем после изнасилования - причины, диагностика, лечение
- Распространенность обрезаний у женщин. Типы женских обрезаний
- Причины обрезания у женщин
- Осложнения женского обрезания - физические, психологические
- Законность женского обрезания. Тактика врача
- Клиника кариотипа 46XY у девочек и женщин
- Генетика нормального полового развития
- Генетический контроль половой дифференцировки - половые хромосомы